使用无水烃溶剂制备助催化剂化合物的方法与流程
[相关申请的交叉引用]本申请要求享有基于2019年5月17日提交的韩国专利申请第10-2019-0058294号的优先权的权益,其全部内容通过引用合并于此。[
技术领域:
]本发明涉及使用无水烃溶剂制备助催化剂化合物的方法,以及由此制备的助催化剂化合物。
背景技术:
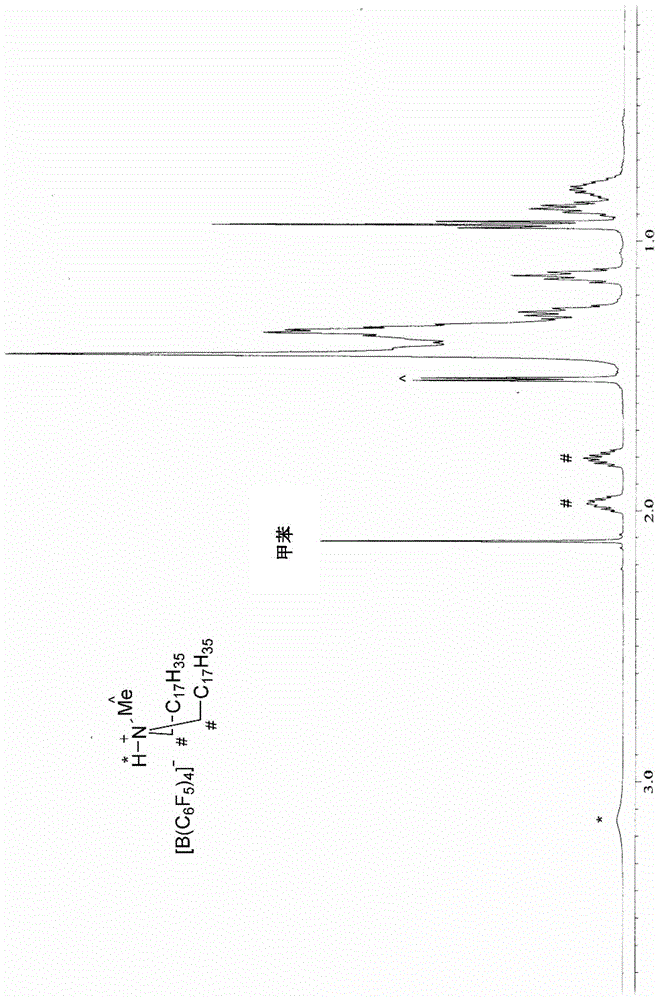
:聚烯烃(po)是主要使用齐格勒-纳塔催化剂制备的聚合物。经常用于制备聚烯烃的常规多相多位点齐格勒-纳塔催化剂已被均相单位点催化剂取代。随着kaminski偶然发现甲基铝氧烷(mao)而开始使用均相单位点催化剂,开始时,已经开发了基于zr的茂金属催化剂(zr-basedmetallocenecatalysts)、基于ti的半茂金属(ti-basedhalf-metallocenes)和具有非环戊二烯基配体的后茂金属(post-metallocenes)催化剂。与基于zr的茂金属催化剂相比,基于ti的半茂金属具有在用于商业生产聚烯烃弹性体的乙烯/α-烯烃共聚反应中显示出更高的α-烯烃混合能力的特性,其典型实例包括陶氏公司(dowco.)在20世纪90年代初期开发的[me2si(η5-me4c5)ntbu]ticl2(约束几何构型催化剂(constrained-geometrycatalyst),cgc)。随着cgc的各种特性逐渐被人们所了解,在学术界和工业界已经积极地进行了合成其衍生物并将其用作聚合催化剂的努力。该方法可以包括改性载体配体,引入其他各种桥和氮取代基代替硅桥,引入氧(oxido)配体代替cgc的胺配体等。另外,作为迄今为止公知的典型金属化合物,已经引入了cgc结构的硅桥,磷,亚乙基或亚丙基,甲叉和甲撑桥等。然而,如果将其用于乙烯聚合或乙烯与α-烯烃的共聚,则与cgc相比时,在聚合活化度或共聚性能方面未显示优异的结果。同时,此类化合物需要使用助催化剂的活化步骤,并且由此可以显示出作为聚合反应催化剂的活性。可以使用基于有机硼酸盐的化合物等作为助催化剂,并且如果通过常规制备方法制备助催化剂,则在制备过程中使用水,并且水分可能残留在助催化剂中,在助催化剂用作活化剂并与过渡金属化合物反应时,这可能会抑制对水脆弱的金属配合物的活性,并且可能会降低作为催化剂的性能。因此,仍然需要开发可以不抑制过渡金属化合物的催化剂活性的助催化剂化合物的制备方法。[现有技术文献][专利文献]韩国注册专利第10-1271055号技术实现要素:技术问题本发明的一个目的是提供一种新颖的使用无水烃溶剂制备由式1表示的助催化剂化合物的制备方法。本发明的另一个目的是提供通过所述制备方法制备的助催化剂化合物和包括该助催化剂化合物的催化剂组合物。技术方案本发明提供了一种制备由下式1表示的化合物的方法,该方法包括:在无水烃溶剂的存在下使由下式2表示的化合物与由下式3表示的化合物反应的步骤:[式1][l-h]+[z(a)4]-[式2][m]+[z(a)4]-[式3][l-h]+[x]-其中,l是含有氮、硫或磷的路易斯碱,z是第13族中的元素,各a独立地为6至20个碳原子的芳基或1至20个碳原子的烷基,其中一个或多个氢原子可被取代基取代,a的取代基是卤素、1至20个碳原子的烃基、1至20个碳原子的烷氧基、或6至20个碳原子的芳氧基,m是第1族中的金属,以及x是卤素元素。有益效果如果根据本发明的制备方法制备助催化剂,并且将助催化剂用于主催化剂化合物的活化反应中,则可以有效提高主催化剂化合物(特别是铪化合物)的催化剂活性,并且进一步地,可以有效地进行单体的聚合反应。此外,为了在商业化过程中确保技术竞争力和成本价格竞争力,要求催化剂的活性高,要求催化剂的用量减少,要求催化剂产物的可再现性优异,并且要求确保稳定性。通过使用根据本发明的制备方法制备的助催化剂可以制备具有显著改善的活性、可再现性和稳定性的催化剂组合物。附图说明图1显示了根据本发明制备的助催化剂[(c18h37)2n(h)me]+[b(c6f5)4]-的1hnmr波谱。图2显示了通过常规制备方法制备的助催化剂[(c18h37)2n(h)me]+[b(c6f5)4]-的1hnmr波谱。具体实施方式在下文中,将更详细地解释本发明以帮助理解本发明。将理解的是,在本发明的说明书和权利要求中使用的词语或术语不应被解释为常用词典中定义的含义。将进一步理解的是,应当基于发明人可以适当地定义词语或术语的含义以最好地解释本发明的原则,将词语或术语解释为具有与本发明的技术思想中的含义一致的含义。在合成助催化剂的常规方法中,使用在有机溶剂中具有低溶解度的反应物(例如,[k][b(c6f5)]),并且在分离过程中产生在有机溶剂中具有低溶解度的副产物(例如,kcl)。因此,在合成和分离过程中必须使用水。在这种方法中,尽管制备助催化剂后在高温/低压条件下除去了水,但助催化剂中不可避免地残留了一定量或更多的水分,并且水分通过与催化剂化合物的反应起到降低催化剂活性的催化剂毒物的作用。特别地,如果如上所述制备包含第13族元素(如硼)的助催化剂,并且水分残留在助催化剂中,则通过与对水分脆弱的主催化剂反应而显著显示出降低催化剂活性的缺陷。为了解决这种缺陷,在本发明中,在合成和分离助催化剂的整个过程中使用无水烃溶剂制备助催化剂,以使水不残留在助催化剂中,并且由此制备的本发明的助催化剂完全不含水分,与少量的主催化剂有效反应以显示出优异的催化剂活性,并显著改善了催化剂的稳定性。特别地,本发明提供了一种制备为助催化剂的由下式1表示的化合物的方法,其特征在于,包括在无水烃溶剂的存在下使由下式2表示的化合物与由下式3表示的化合物反应的步骤:[式1][l-h]+[z(a)4]-[式2][m]+[z(a)4]-[式3][l-h]+[x]-其中,l是含有氮、硫或磷的路易斯碱,z是第13族中的元素,各a独立地为6至20个碳原子的芳基或1至20个碳原子的烷基,其中一个或多个氢原子可被取代基取代,a的取代基是卤素、1至20个碳原子的烃基、1至20个碳原子的烷氧基、或6至20个碳原子的芳氧基,m是第1族中的金属,以及x是卤素元素。特别地,考虑到通过使用无水烃溶剂来解决现有技术的缺陷以及改善作为助催化剂的活性,z可以优选为硼。另外,在上式中,l是含氮的路易斯碱,z是硼,并且各a可以独立地是6至20个碳原子的芳基,其中一个或多个氢原子可以被取代基取代,以及a的取代基可以是卤素或1至20个碳原子的烃基。例如,在式1或式2中包含的[z(a)4]-可以是选自四(苯基)硼酸根、四(五氟苯基)硼酸根、四[3,5-双(三氟甲基)苯基]硼酸根及其衍生物中的一种或多种,但不限于此。通过本发明的制备方法制备的由式1表示的化合物在1hnmr(500mhz,c6d6和标准物质tms)波谱中在1.60ppm至2.10ppm的范围内显示至少两个峰,更特别地,在1.75ppm至1.90ppm的范围和在1.90ppm至2.00ppm的范围的每一个中显示至少一个峰。特别地,在本发明中,如上所述通过使用无水烃溶剂防止水被包括在制备过程中,并且由此制备的由式1表示的化合物在1hnmr波谱中在1.75ppm至1.90ppm的范围和在1.90ppm至2.00ppm的范围的每一个中显示至少一个峰。连接到在l中包含的氮、硫或磷的α-碳上的质子显示不同的峰。例如,如果由式1表示的化合物是[(c18h37)2n(h)me]+[b(c6f5)4]-,则在1hnmr波谱中,在nch2中存在的两个质子显示出各自的峰。这是因为在通过本发明的制备方法制备的助催化剂中水分子没有配位,并且显示出清晰的两个峰,并且由于本发明的助催化剂不包含水分,因此显示的峰是固有峰。相反,在通过相关技术的制备方法制备的助催化剂中,如果通过相同条件测量1hnmr波谱,由于剩余的水分子在nch2中配位,因此仅在1.75ppm至2.00ppm的范围内显示了一个宽的单峰,而没有在1.75ppm至1.90ppm的范围和在1.90ppm至2.00ppm的范围显示不同的峰。在本发明中,通过所述制备方法制备的由式1表示的化合物的实例可以是,如果z是硼,二(十八烷基)甲基铵四(五氟苯基)硼酸盐[(c18h37)2n(h)me]+[b(c6f5)4]-、二(十八烷基)甲基铵四(苯基)硼酸盐、二(十八烷基)甲基铵四[3,5-双(三氟甲基)苯基]硼酸盐四(苯基)硼酸盐、三乙基铵四(苯基)硼酸盐、三丁基铵四(苯基)硼酸盐、三甲基铵四(苯基)硼酸盐、三丙基铵四(苯基)硼酸盐、三甲基铵四(对甲苯基)硼酸盐、三甲基铵四(邻,对二甲基苯基)硼酸盐、三丁基铵四(对三氟甲基苯基)硼酸盐、三甲基铵四(对三氟甲基苯基)硼酸盐、三丁基铵四(五氟苯基)硼酸盐、n,n-二乙基苯铵四(苯基)硼酸盐、n,n-二乙基苯铵四(苯基)硼酸盐、n,n-二乙基苯铵四(五氟苯基)硼酸盐、二乙基铵四(五氟苯基)硼酸盐、三苯基鏻四(苯基)硼酸盐、三甲基鏻四(苯基)硼酸盐、三甲基铵四(苯基)硼酸盐、三丙基铵四(苯基)硼酸盐、三甲基铵四(对甲苯基)硼酸盐、三丙基铵四(对甲苯基)硼酸盐、三乙基铵四(邻,对二甲基苯基)硼酸盐、三甲基铵四(邻,对二甲基苯基)硼酸盐、三丁基铵四(对三氟甲基苯基)硼酸盐、三甲基铵四(对三氟甲基苯基)硼酸盐、三丁基铵四(五氟苯基)硼酸盐、n,n-二乙基苯铵四(苯基)硼酸盐、n,n-二乙基苯铵四(苯基)硼酸盐、n,n-二乙基苯铵四(五氟苯基)硼酸盐、二乙基铵四(五氟苯基)硼酸盐、三苯基鏻四(苯基)硼酸盐、三苯基碳鎓四(对三氟甲基苯基)硼酸盐、三苯基碳鎓四(五氟苯基)硼酸盐、或其组合,以及如果z是铝,例如三乙基铵四(苯基)铝、三丁基铵四(苯基)铝、三甲基铵四(苯基)铝、三丙基铵四(苯基)铝、三甲基铵四(对甲苯基)铝、三丙基铵四(对甲苯基)铝、三乙基铵四(邻,对二甲基苯基)铝、三丁基铵四(对三氟甲基苯基)铝、三甲基铵四(对三氟甲基苯基)铝、三丁基铵四(五氟苯基)铝、n,n-二乙基苯铵四(苯基)铝、n,n-二乙基苯铵四(苯基)铝、n,n-二乙基苯铵四(五氟苯基)铝、n,n-二乙基铵四(五氟苯基)铝、二乙基铵四(五氟苯基)铝、三苯基鏻四(苯基)铝、三甲基鏻四(苯基)铝、三乙基铵四(苯基)铝、三丁基铵四(苯基)铝或其组合,但不限于此。在本发明中,烃溶剂可以使用选自丁烷、戊烷、新戊烷、己烷、环己烷、甲基环己烷、庚烷、辛烷、苯、甲苯、二甲苯、乙苯、二氯甲烷和氯仿的一种或多种,但不限于此,在本
技术领域:
中使用的所有烃溶剂都可以以无水形式使用。在本发明中,由式2表示的化合物与由式3表示的化合物的反应可以在0至50℃的温度下进行,优选地,在10至40℃的温度下进行,或者在15至30℃的温度下进行。另外,所述反应可以进行15分钟至5小时,或30分钟至3小时,或30分钟至1小时。另外,在所述反应中,由式2表示的化合物与由式3表示的化合物的摩尔比可以为1:1至5:1或1:1至2:1。另外,本发明提供了通过所述制备方法制备的由式1表示的化合物。如上所述,在本发明中制备的由式1表示的化合物在1h-nmr波谱中显示固有峰,并且在活化用于聚合烯烃的催化剂时显示出高催化剂活性和催化剂稳定性。本发明的由式1表示的化合物是助催化剂化合物,并且可以以包括该化合物的助催化剂组合物的形式使用。所述“组合物”包括包含在相应组合物中的材料的混合物以及由相应组合物的材料形成的反应产物和分解产物。所述助催化剂组合物,除了本发明的由式1表示的化合物之外,还可以包含本
技术领域:
中常用的助催化剂,并且可以特别地进一步包含选自以下式4和5中的一种或多种:[式4]-[al(ra)-o]m-[式5]d(ra)3其中,各ra独立地是卤素基团,1至20个碳原子的烃基,或1至20个碳原子的卤素取代的烃基,m是2或更大的整数,以及d是铝或硼。由式4表示的化合物没有特别限制,只要使用烷基铝氧烷即可。其优选实例可包括甲基铝氧烷、乙基铝氧烷、异丁基铝氧烷、丁基铝氧烷等,更优选甲基铝氧烷。由式5表示的化合物没有特别限制,并且其优选实例可以包括三甲基铝、三乙基铝、三异丁基铝、三丙基铝、三丁基铝、二甲基氯化铝、三异丙基铝、三仲丁基铝、三环戊基铝、三戊基铝、三异戊基铝、三己基铝、三辛基铝、乙基二甲基铝、甲基二乙基铝、三苯基铝、三对甲苯基铝、二甲基甲氧基铝、二甲基乙氧基铝、三甲基硼、三乙基硼、三异丁基硼、三丙基硼、三丁基硼等,更优选选自三甲基铝、三乙基铝和三异丁基铝。所述助催化剂组合物可以用作过渡金属化合物的催化剂活化剂,并且过渡金属化合物和助催化剂组合物可以通过载体以负载型使用。氧化硅或氧化铝可用作载体。在本发明中,所述助催化剂组合物可以用于包含有机金属化合物作为主催化剂的催化剂组合物中。作为在催化剂组合物中用作主催化剂的有机金属化合物,可以使用所有有机金属催化剂,只要中心金属是ti、hf或zr,并且结构是茂金属、半茂金属或后茂金属即可,而没有限制。在催化剂组合物中包含的由式1表示的化合物与主催化剂的相互作用是顺畅的,并且作为主催化剂的有机金属化合物被活化以有效地实现催化剂活性,并因此可以改善烯烃单体的聚合反应的效率。所述催化剂组合物可以通过使本发明的由式1表示的化合物与有机金属化合物接触的步骤来制备。由式1表示的化合物与有机金属化合物的摩尔比可以为1:0.05至1:2,特别是1:0.1至1:1,或1:0.2至1:1。如果由式1表示的化合物的量小于上述范围,则助催化剂的量相对小于主催化剂的量,并且茂金属化合物的活化可能不完全地实现,从而产生降低催化剂组合物的活性的缺陷,而如果由式1表示的化合物的量大于上述范围,则可以完全实现茂金属化合物的活化,但是由于残留了过量的助催化剂,因此可以出现催化剂组合物的单位成本不经济以及聚合物的纯度降低的缺陷。作为在组合物的制备过程中使用的反应溶剂,可以使用基于烃的溶剂,例如,戊烷,己烷和庚烷,以及芳族溶剂,例如,苯和甲苯,但不限于此,并且可以使用本
技术领域:
中使用的所有溶剂。另外,由式1表示的化合物和有机金属化合物可以以在载体中的负载形式使用。作为载体,可以使用氧化硅或氧化铝。另外,本发明提供了一种烯烃聚合物的制备方法,包括在所述催化剂组合物的存在下使烯烃单体聚合的步骤。使用所述催化剂组合物的最优选的制备方法是溶液法,并且如果该组合物与无机载体(如氧化硅)一起使用,则也可以应用淤浆法或气相法。用于活化的催化剂组合物可以在溶解或稀释于5至12个碳原子的脂族烃溶剂(如戊烷、己烷、庚烷、壬烷、癸烷、其异构体),芳族烃溶剂(如甲苯和苯),或被氯原子取代的烃溶剂(如二氯甲烷和氯苯)中之后注入,它们适用于烯烃聚合工艺。使用的溶剂优选通过用少量的烷基铝处理除去少量的水或空气(其起催化剂毒物的作用)后使用,并且可以通过进一步使用助催化剂来使用。在本发明中,烯烃单体可以是选自乙烯、丙烯、1-丁烯、1-戊烯、4-甲基-1-戊烯、1-己烯、1-庚烯、1-辛烯、1-癸烯、1-十一碳烯、1-十二碳烯、1-十四碳烯、1-十六碳烯和1-二十碳烯的一种或多种,但不限于此。特别地,根据烯烃单体的类型,烯烃聚合物可以是烯烃均聚物、烯烃/α-烯烃共聚物,优选乙烯/α-烯烃共聚物。在这种情况下,本领域技术人员可以根据烯烃聚合物的用途、目的等适当选择作为共聚单体的α-烯烃单体的量,并且可以为约1至约99mol%。实施例在下文中,将参考实施例更具体地说明本发明。然而,这些实施例用于说明本发明,并且本发明的范围不限于此。试剂和实验条件所有实验均使用标准手套箱和schlenk技术在惰性气氛下进行。甲苯、己烷和thf在由二苯甲酮羰自由基(benzophenoneketyl)蒸馏后使用。聚合反应中使用的甲基环己烷(无水级)从东京化学工业公司(tci)购买并使用na/k合金纯化。hfcl4(升华级)购自streme,并按原样使用。乙烯/丙烯混合气体在bomb反应器(2.0l)中在三辛基铝(0.6m,在甲基环己烷中)上纯化。使用joelecz600仪器测量1hnmr(600mhz)和13cnmr(150mhz)波谱,并在韩国亚洲(ajou)大学分析中心进行元素分析。使用配备ri检测器和两根色谱柱(varian,plgelmixed-b7.5x300mm)的pl-gpc220系统,在160℃下通过1,2,4-三氯苯获得gpc数据。助催化剂的制备制备实施例1过量的[k]+[b(c6f5)4]-(0.633g,0.881mmol,纯),和[(c18h37)2n(h)me]+[cl]-(0.404g,0.705mmol)在甲苯(无水,10ml)中的溶液在25℃的手套箱中反应1小时。在硅藻土上过滤后,使用真空管线除去溶剂。将残余物溶于甲基环己烷(4ml),然后再次在硅藻土上过滤。通过除去溶剂,产生黄色油状化合物,其无需另外分离即可使用(0.797g,93%)。分析1hnmr波谱,结果示于图1中。比较制备实施例1将k+[b(c6f5)4]-(3g,4.177mmol,纯)和[(c18h37)2n(h)me]+[cl]-(2.511g,4.386mmol)在甲苯(130ml)和水(120ml)的存在下在室温下反应1小时。向其中加入200ml的na2co3饱和溶液,并使用分液漏斗从混合物中除去水层。加入100ml的na2co3饱和溶液并洗涤,并且添加100ml的蒸馏水并另外洗涤。注入mgso4以除去水,并将所得产物过滤。使用旋转蒸发仪从滤液中除去溶剂,得到黄色油状化合物(4.16g,82%)。在制备实施例1的1hnmr波谱中,观察到两个键合到α-碳(nch2)上的质子,分别在1.97ppm和1.80ppm处(图1)。-1hnmr(c6d6):δ3.15(br,h,nh),1.97(m,2h,nch2),1.80(m,h,nch2),1.51(d,j=6.0hz,3h,nch3),1.45-1.29(m,48h),1.26(五重峰,j=7.2hz,4h),1.13(五重峰,j=7.2hz,4h),0.94(t,j=7.8hz,6h),0.88(五重峰,j=7.8hz,4h),0.81(m,4h)ppm.19fnmr(c6d6):δ-132.09,-161.75,-165.98.相反,根据含有水的比较制备实施例1的1hnmr波谱,观察到nch2的质子为约1.89ppm的单个宽信号(图2)。也就是说,可以发现本发明中制备的无水[(c18h37)2n(h)me]+[b(c6f5)4]-助催化剂是高纯度的化合物,不含痕量的可以与胺基配位的水,与通过现有技术的方法制备的助催化剂不同。烯烃聚合物的制备(1)催化剂组合物的制备实施例1[式a]通过将制备实施例1的助催化剂(2.5μmol),由式a表示的主催化剂(2.5μmol)和甲基环己烷(2.5ml)混合来制备催化剂组合物。实施例2和3,以及比较实施例1至3除了如表1中改变助催化剂和主催化剂的注入条件之外,以与实施例1相同的方法制备催化剂组合物。[表1](2)烯烃单体的聚合将30ml的己烷和100g的甲基环己烷注入到90℃的高压反应器中。在将催化剂组合物注入反应器之后,立即以20巴的压力注入乙烯。在95℃至100℃的温度下进行聚合40分钟,并排出剩余的气体。实验实施例1基于实施例和比较实施例的结果,通过以下方法分析催化剂的特性。(1)催化剂活性(吨/molhf/小时)基于单位小时(hr),将催化剂活性计算为所产生的聚合物的重量(吨)/所使用的催化剂含量(hfmol)的比率。[表2]催化剂活性(吨/hfmol/hr)实施例128实施例235实施例335比较实施例14.7比较实施例23比较实施例3无活性如表2中所示,证实了使用根据本发明的制备方法制备的助催化剂的实施例1至3在烯烃聚合期间显示出比比较实施例1至3更高的催化剂活性。特别地,证实了,制备实施例1的助催化剂尽管如实施例3中那样以1μmol的少量使用,但显示出高催化剂活性,但是在使用比较制备实施例1中的助催化剂的比较实施例中,催化剂活性显著降低,特别是在比较实施例3中,由于比较制备实施例1的助催化剂中所含的水分而使催化剂失活,根本没有显示出催化剂活性。(2)催化剂活性随时间流逝的变化(催化剂稳定性)为了评估催化剂溶液的稳定性,制备了实施例和比较实施例的催化剂组合物,将其静置如下表3所示的一定时间,并用于制备烯烃聚合物。评价每种催化剂的活性,并示于表4。[表3]静置时间实施例11天实施例23天实施例37天比较实施例16小时比较实施例212小时比较实施例31天[表4]如表4所示,使用制备实施例1的助催化剂的实施例的催化剂稳定性优异,并且在经过一段时间后几乎原样保持主催化剂的活性,但是使用比较制备实施例1的助催化剂的比较实施例的催化剂稳定性低,并且发现随着时间的流逝催化剂活性迅速降低。也即是说,可以证实,如果使用根据本发明的制备方法制备的助催化剂,则主催化剂化合物的初始催化剂活性显示为高,催化剂稳定性也优异,并且经过一段时间后仍保持高催化剂活性。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1