热诱导交联的酚酞基聚苯并噁唑气体分离膜材料及其制备方法
1.本发明属于膜分离领域,具体地,涉及热诱导交联的酚酞基聚苯并噁唑气体分离膜材料,热诱导交联的酚酞基聚苯并噁唑气体分离膜材料的制备方法,以及由该制备方法制得的酚酞基聚苯并噁唑气体分离膜材料。
背景技术:
2.在过去二十年中,开发用于气体分离的新型聚合物引起了科研工作者的极大兴趣。聚酰亚胺具有较高的气体选择性、优异的热稳定性、较好的机械性能和良好的成膜性,被认为是一种很有前景的气体分离材料。但是,传统的聚酰亚胺膜气体渗透性低,而且在分离含有较高浓度可溶气体(co2和ch4等)时易发生塑化。塑化是指随着进料侧压力的增加,聚合物基体中的某些组分发生溶胀,聚合物链间的流动性增加,表现出的结果是随着上游压力的增加,气体渗透性增加,选择性降低。为了改善气体分离性能,科研工作者采取了很多方法来改变聚酰亚胺的物理和化学性质。通过在聚合物主链引入刚性的大侧基cardo基团可以使聚合物链的堆积密度降低,聚合物的自由体积增加,气体渗透性也增加。
3.cardo型聚合物是含有环状结构单元的聚合物,并且这些环状结构单元的一个原子处在主链上。将双苯基芴基引入到聚酰亚胺主链上,苯芴基聚合物一般具有优异的热稳定性和较高的溶解度,结果表明引入cardo型大侧基会降低主链的旋转运动性,导致聚合物的t
g
大大增加;同时,破坏分子链的规整排列,分子链间距增加,聚合物膜的气体渗透性和选择性都有所提高(s.kazama,et al.journal of membrane science.2002,(207):91
‑
104)。
4.通过合成含叔丁基环己基双苯基的二胺单体,与不同的二酐单体反应生成聚酰亚胺。结果表明在聚酰亚胺的主链上引入大位阻cardo侧基结构,阻碍了分子链的规整排列,从而极大的改善聚合物溶解性和可加工性,降低分子链的堆积密度,提高聚合物的自由体积(der
‑
jang liaw,et al.macromolecular chemistry and physics.2000,(201):1887
‑
1893)。
5.通过对聚酰亚胺交联可以有效的解决膜材料的塑化问题,交联也可以提高聚合物的热稳定性和化学稳定性。传统的交联方法主要有热交联、化学交联和uv交联等。化学交联需要在反应体系中引入二胺或二醇小分子交联剂,发生交联反应形成酰胺键或酯键,在酸性条件下易发生水解。热诱导交联可以在t
g
以下发生,在高纯度空气氛围下或者高纯氮气氛围下的高温炭化炉中进行。当在高纯度空气氛围下聚合物内酯环分解成自由基,自由基与氧气分子形成过氧自由基,过氧自由基捕捉相邻聚合物的自由基或氢原子形成过氧化物,过氧化物极易裂解成新的自由基,新的自由基与周围链上的自由基反应形成交联结构。当在高纯度氮气氛围下聚合物内酯环分解成自由基,自由基捕捉氢原子或者相邻聚合物的苯环自由基形成苯环或者联苯交联结构。通过缩聚反应将内酯环结构引入到聚酰亚胺中,将制备完成的膜放入高纯度空气氛围的碳化炉中低于t
g
热处理,结果表明交联使得聚合物
的刚性和链间距增加,聚合物的渗透率和选择性增加,同时具有很好地抗塑化性(caili zhang,et al.journal of membrane science.2018,(546):90
‑
99)。酚酞基聚醚酮(pek
‑
c)在高纯氮气下进行热处理,内酯环分解发生热诱导链间交联形成联苯键,交联之后刚性的交联网络形成,自由体积增加,使得膜的抗塑化性和渗透性提高(ruisong xu,et al.journal of membrane science.2019,(586):306
‑
317)。
6.聚苯并噁唑(pbo)是一类主链含有苯并噁唑稠杂环的芳香族聚合物。含邻羟基的聚酰亚胺发生高温热重排(tr)反应形成芳杂环结构,pbo上的刚性芳香环结构使主链上亚苯基和苯并噁唑环之间的旋转具有高的扭转能垒,破坏分子链的对称性和规整性,同时这种空间结构上的重排可以有效提高聚合物骨架的刚性,使聚合物膜具有高渗透性和高选择性,而且还具有超强的热稳定性和化学稳定性以及超高的拉伸强度。使用含cardo基团的二胺单体制备pbo气体分离膜,结果表明刚性大体积cardo基团的引入会改变模腔自由体积的尺寸,这种空腔分布加快了气体分子的传输速率,提高了膜的气体渗透性(y.f.yeong.,journal of membrane science,2012,397,56)。
7.酚酞基的典型结构为三苯甲醇邻苯甲酸内酯结构,大量苯环的存在使这类聚合物具有优异的热稳定性,内酯环结构的存在又使这类聚合物具有较好的溶解性。侧基的芳杂环结构降低聚合物链的密度,增大自由体积,有利于提高气体渗透性。因此希望制备一种高性能的酚酞基聚苯并噁唑气体分离膜,先合成含大体积酚酞基的聚酰亚胺,可以对这类含内酯环的聚合物进行热诱导交联处理,经交联之后的膜材料再继续升高温度热重排处理形成pbo膜材料,这种新型膜材料结合了酚酞基cardo结构、热重排苯并噁唑结构以及热诱导交联结构的特点。
技术实现要素:
8.本发明的目的是针对现有pbo分离膜制备过程中的一些不足,选用酚酞、荧光素等含有cardo大侧基的单体,价格低,易获取,最重要的是采用新的硝化方法,反应条件温和,硝化产品容易控制,避免了柱层析等提纯过程,获得聚酰亚胺膜之后在t
g
以下进行热诱导交联处理,既获得了优异的三维网状交联结构,又避免了聚合物膜结构的破坏。最后升高温度膜材料发生热重排反应可以高效率工业化生产高性能的pbo气体分离膜材料。
9.为了实现上述目的,本发明的构思是对酚酞类单体进行硝化、还原合成酚酞基二胺,再与二酐发生亚胺化反应合成聚酰亚胺,再用这种含酚酞基内酯环的聚酰亚胺制成膜,在高纯度的空气氛围或者氮气氛围下的碳化炉t
g
以下热诱导交联处理,继续升高温度高温热处理使前驱体发生热重排反应转化成聚苯并噁唑膜用于气体分离。
10.具体地,本发明提供热诱导交联的酚酞基聚苯并噁唑气体分离膜,其结构式为式i或式ii所示(标记的1和2为热诱导交联时自由基的进攻位点):
[0011][0012]
其中,r1、r2、r3各自独立地为h或c1‑
c4烷基;
[0013]
m1、m2和n表示重复单元的数量,在20到1000之间;
[0014]
ar为二酐酐基的连接单元,选自以下结构中的任意一种:
[0015][0016]
其中,弯折线表示二酐酐基和ar的连接键;
[0017]
所述二酐酐基衍生自二酐单体,所述二酐单体选自4,4'
‑
(六氟异丙烯)二酞酸酐、3,3’,4,4
’‑
二苯酮四酸二酐或二苯醚四酸二酐。
[0018]
根据本发明,所述酚酞基聚苯并噁唑气体分离膜选自以下任选一种:
[0019]
(i)式i中r1=h、r2=h、r3=h;
[0020]
(ii)式i中r1=h、r2=ch3、r3=ch3;
[0021]
(iii)式ii中r1=h、r2=h、r3=h;
[0022]
(iv)式ii中r1=h、r2=ch3、r3=ch3。
[0023]
根据本发明,当式i中r1=h、r2=h、r3=h时,所述酚酞基聚苯并噁唑气体分离膜是由单体(a)酚酞发生硝化、还原、亚胺化、热诱导交联、热重排反应合成的pbo聚合物;
[0024]
(a)酚酞。
[0025]
根据本发明,当式i中r1=h、r2=ch3、r3=ch3时,所述酚酞基聚苯并噁唑气体分离膜是由单体(b)邻甲酚酞发生硝化、还原、亚胺化、热诱导交联、热重排反应合成的pbo聚合物;
[0026]
(b)邻甲酚酞。
[0027]
根据本发明,当式ii中r1=h、r2=h、r3=h时,所述酚酞基聚苯并噁唑气体分离膜是由单体(c)荧光素发生硝化、还原、亚胺化、热诱导交联、热重排反应合成的pbo聚合物;
[0028]
(c)荧光素。
[0029]
根据本发明,当式ii中r1=h、r2=ch3、r3=ch3时,所述酚酞基聚苯并噁唑气体分离膜是由单体(d)4,4'
‑
二甲基荧光素发生硝化、还原、亚胺化、热诱导交联、热重排反应合成的pbo聚合物;
[0030]
(d)4,4'
‑
二甲基荧光素。
[0031]
本发明还提供热诱导交联的酚酞基聚苯并噁唑气体分离膜材料的制备方法,该方法包括以下步骤:
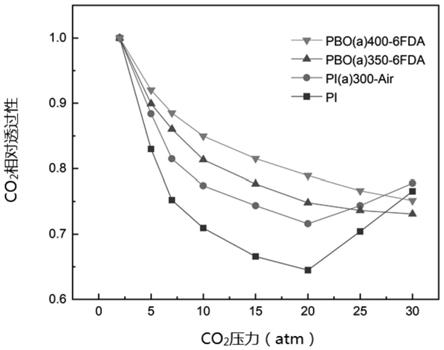
[0032]
(1)二硝单体的合成
[0033]
将单体溶于乙酸溶液中,搅拌均匀,加入六水合硝酸钇,继续搅拌,加入过量的冰冷的去离子水,析出沉淀,过滤并用冰冷的去离子水冲洗,得到的滤渣在真空烘箱干燥;所述单体选自酚酞、荧光素、4,4'
‑
二甲基荧光素或邻甲酚酞;
[0034]
(2)二胺单体的合成
[0035]
称取步骤(1)得到的二硝单体,溶于四氢呋喃和乙醇混合溶剂中,加入pd/c催化剂,对反应装置抽真空、通入h2进行反应,反应结束后,冷却至常温,过滤除去催化剂pd/c,趁热不断搅拌缓慢加入适量的去离子水到浊点,冷却结晶,析出固体颗粒,过滤,真空干燥;
[0036]
(3)聚酰亚胺的合成
[0037]
取二酐单体溶于溶剂中,在低温冰浴和氮气保护下搅拌,批量加入步骤(2)得到的二胺单体,持续搅拌反应,直到形成高粘度的聚酰胺酸溶液,加入乙酸酐和无水吡啶,室温
下继续反应,将粘度很高的聚酰亚胺溶液倒在甲醇溶液中沉淀,过滤时用甲醇洗涤,除去残留溶剂,最后将得到的聚酰亚胺滤渣在真空烘箱中干燥;所述二酐单体选自4,4'
‑
(六氟异丙烯)二酞酸酐(6fda)、3,3’,4,4
’‑
二苯酮四酸二酐(btda)或二苯醚四酸二酐(odpa);
[0038]
(4)聚酰亚胺致密膜的制备
[0039]
取步骤(3)制得的聚合物粉末溶解于和步骤(3)相同的溶剂中,搅拌直至完全溶解,用聚四氟乙烯滤膜过滤,将过滤之后的溶液制为聚酰亚胺致密膜;最后将聚合物膜真空干燥,除去残余溶剂,得到的聚合物膜的膜厚在70~80μm之间;
[0040]
(5)热诱导交联处理致密膜
[0041]
将步骤(4)干燥好的聚酰亚胺膜置于碳化炉中,在空气氛围或者氮气氛围下,程序升温至250
‑
300℃并保温,得到所述热诱导交联处理致密膜;所述程序升温至250
‑
300℃并保温的条件优选包括:以4~6℃/min的升温速率分别升温至250℃、275℃和300℃,保温时间为1~3h;
[0042]
(6)热重排聚合物膜的制备
[0043]
分别将三个温度下保温1~3h热诱导交联处理之后的致密膜置于碳化炉中,在氮气氛围下程序升温至350
‑
450℃并保温,得到所述热重排聚合物膜;
[0044]
所述程序升温至350
‑
450℃并保温的条件优选包括:以4~6℃/min的升温速率分别升温至350℃、400℃和450℃,保温时间为1~2h。
[0045]
根据本发明,优选地,步骤(1)包括:
[0046]
称取一定量的单体,溶于乙酸溶液中搅拌均匀,加入相同摩尔质量的六水合硝酸钇,继续搅拌1~2h,加入过量的冰冷的去离子水,有大量的沉淀析出,搅拌1~2h,过滤时用冰冷的去离子水冲洗,得到的滤渣置于50~70℃真空烘箱干燥。
[0047]
根据本发明,优选地,步骤(2)包括:
[0048]
称取一定量的步骤(1)制得的二硝单体,溶于四氢呋喃和乙醇按1:1~2比例混合的溶剂中,加入单体量10%的pd/c催化剂,对反应装置抽真空、通入h2,50~70℃下反应10~12h,反应结束后,冷却至常温,过滤除去催化剂pd/c,趁热不断搅拌缓慢加入适量的去离子水到浊点,冷却结晶,析出固体颗粒,过滤,烘箱50~70℃下真空干燥。
[0049]
根据本发明,优选地,步骤(3)包括:
[0050]
取一定量的二酐单体溶于溶剂中,在低温冰浴和氮气保护下搅拌20~40min,批量加入步骤(2)制得的二胺单体,溶液的浓度为18~22wt%,持续搅拌反应10~14h,直到形成高粘度的聚酰胺酸溶液,加入一定量的乙酸酐和无水吡啶,撤掉冰浴,室温下继续反应20~28h,将粘度很高的聚酰亚胺溶液倒在甲醇溶液中沉淀,过滤时用甲醇洗涤,除去残留溶剂,最后将得到的聚酰亚胺滤渣70~80℃在真空烘箱中干燥20~28h;所述溶剂选自n
‑
甲基
‑2‑
吡咯烷酮、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺。
[0051]
根据本发明,优选地,步骤(4)包括:
[0052]
取一定量的聚合物粉末溶解于和步骤(3)相同的溶剂中,搅拌直至完全溶解,用0.45μm的聚四氟乙烯滤膜过滤,将过滤之后的溶液注入平整的表面皿中,然后将装入自封袋,室温下在平整的台子上放置3~4天,得到厚度均匀,表面光滑平整聚酰亚胺致密膜。最后将聚合物膜放在180~200℃的烘箱中真空干燥20~28h,除去残余溶剂,最后得到的聚合物膜的膜厚在70~80μm之间。
[0053]
本发明还提供由上述制备方法制得的热诱导交联的酚酞基聚苯并噁唑气体分离膜材料。
[0054]
本发明具有以下优点:
[0055]
本发明选用易获取、价格低廉的含酞结构单体,与六水合硝酸钇发生硝化反应,得到高纯度的二硝产物,反应条件温和,硝化产品容易控制,避免了柱层析等工艺复杂的提纯过程。t
g
以下内酯环分解成自由基发生热诱导交联,既改善了聚合物膜的抗塑化性,又避免了高温交联造成孔隙塌陷的问题。同时由于这类单体含有酚羟基,可以在对聚酰亚胺膜热诱导交联之后,进一步发生热重排反应,形成结构更加扭曲更加刚性的聚苯并噁唑膜。此外,含酞结构的引入使此类聚合物具有热稳定性好、溶解性好和透明度高等优点。将此类pbo膜用于气体分离,气体渗透性得到很大提高,其中co2的通量提高了70多倍。
[0056]
本发明的其它特征和优点将在随后具体实施方式部分予以详细说明。
附图说明
[0057]
通过结合附图对本发明示例性实施方式进行更详细的描述,本发明的上述以及其它目的、特征和优势将变得更加明显。
[0058]
图1为本发明中单体(a)发生硝化、还原、与6fda发生反应合成聚酰亚胺的反应方程式。
[0059]
图2为本发明实施例1中3,3'
‑
二硝基酚酞、3,3'
‑
二氨基酚酞和3,3'
‑
二氨基酚酞与6fda反应合成的聚酰亚胺的1h
‑
nmr谱图(dmso
‑
d6),其中,a为3,3'
‑
二硝基酚酞的1h
‑
nmr谱图(dmso
‑
d6),b为3,3'
‑
二氨基酚酞的1h
‑
nmr谱图(dmso
‑
d6),c为3,3'
‑
二氨基酚酞与6fda发生反应合成的聚酰亚胺的1h
‑
nmr谱图(dmso
‑
d6)。
[0060]
图3为本发明实施例1中聚酰亚胺发生在空气氛围下热诱导交联、热重排反应的流程图。
[0061]
图4为本发明实施例1中制备的聚酰亚胺膜。a:pi膜的柔韧性展示;b:pi膜。
[0062]
图5为本发明实施例1中聚酰亚胺膜在高纯度空气氛围下发生热诱导交联的反应机理图。
[0063]
图6为本发明实施例1中的聚酰亚胺膜分别在250℃、275℃和300℃高纯度空气氛围下发生热诱导交联处理得到的膜,以及300℃热诱导交联处理之后的膜分别在350℃、400℃和450℃发生热重排反应合成的pbo膜,a:pi(a)250
‑
air膜;b:pi(a)275
‑
air膜;c:pi(a)300
‑
air膜;d:pbo(a)350
‑
6fda膜;e:pbo(a)400
‑
6fda膜;f:pbo(a)450
‑
6fda膜。
[0064]
图7为本发明实施例1中的聚酰亚胺未处理膜、300℃高纯空气氛围下热诱导交联膜、350℃热处理的pbo膜和400℃热处理的pbo膜抗塑化性能对比图。
[0065]
图8为本发明实施例1中的最优条件下处理前后聚合物膜的红外光谱对比图。
[0066]
图9为本发明中单体(b)发生硝化、还原、与6fda发生反应合成聚酰亚胺的反应方程式。
[0067]
图10为本发明实施例2中聚酰亚胺膜在高纯度氮气氛围下发生热诱导交联的反应机理图。
[0068]
图11为本发明实施例2中的聚酰亚胺未处理膜、300℃高纯氮气氛围下热诱导交联膜、350℃pbo膜和400℃pbo膜抗塑化性能对比图。
具体实施方式
[0069]
下面将更详细地描述本发明的优选实施方式。虽然以下描述了本发明的优选实施方式,然而应该理解,可以以各种形式实现本发明而不应被这里阐述的实施方式所限制。
[0070]
实施例1
[0071]
单体(a)发生硝化、还原、与6fda发生反应合成聚酰亚胺,反应方程式如图1所示。
[0072]
称取酚酞1.0g,溶于30ml的乙酸中搅拌均匀,再加入1.2g的y(no3)3·
6h2o,反应1h,加入50ml冰冷的去离子水,有大量的黄色沉淀析出,搅拌1h,过滤时用大量的冰冷的去离子水冲洗,得到的滤渣于60℃真空烘箱干燥,产率为89%。所得3,3'
‑
二硝基酚酞的1h
‑
nmr谱图(dmso
‑
d6)如图2的a所示。
[0073]
称取5.35g 3,3'
‑
二硝基酚酞置于100ml三口瓶中,依次加入30ml的thf和20ml的乙醇,搅拌均匀之后加入0.5g的pd/c催化剂,对反应装置抽真空、通入h2,60℃下反应12h,反应结束后,冷却至常温,过滤除去pd/c,趁热不断搅拌缓慢加入适量的去离子水到浊点,冷却结晶,析出固体颗粒,过滤,烘箱60℃下真空干燥。得到白色粉末2.89g。产率为63.38%。所得3,3'
‑
二氨基酚酞的1h
‑
nmr谱图(dmso
‑
d6)如图2的b所示。
[0074]
取6fda 1.78g(444.24g/mol,4mmol)加入100ml的三口烧瓶中,加入12ml dmac在低温冰浴和氮气保护下搅拌30min,分两次加入3,3'
‑
二氨基酚酞1.42g(348.32g/mol,4.08mmol),固含量为21wt%,持续搅拌反应14h,形成高粘度的聚酰胺酸溶液,加入5ml的乙酸酐、5ml的无水吡啶,撤掉冰浴。室温下继续反应24h,将粘度很高的聚酰亚胺溶液倒在甲醇溶液中沉淀,过滤时用甲醇洗涤,除去残留溶剂,80℃烘箱抽真空干燥24h,得到干燥后的聚酰亚胺2.03g。3,3'
‑
二氨基酚酞与6fda发生反应合成的聚酰亚胺的1h
‑
nmr谱图(dmso
‑
d6)如图2的c所示。对于聚酰亚胺的核磁氢谱需要说明的是,在化学亚胺化的过程中由于乙酸酐的存在,导致邻位羟基向邻位乙酸酯基转化。不过这种转化对后处理不会造成影响,因为在250℃左右乙酸酯基又可以转化为羟基。
[0075]
取0.35g干燥好的聚酰亚胺粉末溶解于dmf中,搅拌直至完全溶解,铸膜液固含量32wt%,用0.45μm的ptfe滤膜过滤,将过滤之后的溶液注入平整的表面皿中,然后将其装入自封袋,室温下在平整的台子上放置4天,得到厚度均匀、表面光滑平整的聚酰亚胺致密膜。最后将聚合物膜放在200℃的烘箱中真空干燥24h,除去残余溶剂,最终所得聚合物膜的膜厚在70~80μm之间。聚酰亚胺发生在空气氛围下热诱导交联、热重排反应的流程图如图3所示。
[0076]
将如图4所示的干燥好的聚酰亚胺膜置于碳化炉中,在空气氛围下,以5℃/min的升温速率分别升温至250℃、275℃和300℃,保温时间为2h,得到的热诱导交联膜,分别命名为pi(a)250
‑
air、pi(a)275
‑
air、pi(a)300
‑
air。聚酰亚胺膜在空气氛围下发生热诱导交联的反应机理图如5所示。
[0077]
将热诱导交联处理2h之后的致密膜置于碳化炉中,在氮气氛围下,以5℃/min的升温速率分别升温至350℃、400℃和450℃,保温时间为1.5h,其中300℃下热诱导交联之后处理得到的pbo膜分别命名为pbo(a)350
‑
6fda、pbo(a)400
‑
6fda、pbo(a)450
‑
6fda。
[0078]
所得热诱导交联的酚酞基聚苯并噁唑气体分离膜中,m1、m2、n的值在30至100之间。
[0079]
处理前后的聚合物膜图片对比如图6所示,表1为单体(a)合成的聚酰亚胺膜、300℃空气氛围下热诱导交联处理2h的膜和pbo(a)350
‑
6fda、pbo(a)400
‑
6fda、pbo(a)450
‑
6fda膜气体透过性和气体选择性。
[0080]
表1
[0081][0082]
从表1中可以看出,热诱导交联之后渗透性增加,这主要是因为酚酞基上的内酯环开始分解成自由基形成交联结构,链间距增加。经热重排之后得到的聚苯并噁唑气体分离膜,气体透过性得到了很大的提高,其中450℃处理之后co2的通量高达2169barrer。相比于前驱体聚酰亚胺气体分离膜,热重排聚合物膜π
‑
π堆积程度减少,但由于酰亚胺键转化成了噁唑环,链段刚性增加,使得聚合物膜的气体选择性在400℃时有所增加。
[0083]
从图7可以看出,对于可冷凝气体co2,由于高压下聚合物膜溶胀导致增塑作用,玻璃态聚合物的渗透等温线可能会呈现向上的趋势。当co2的压力高于20atm时,未处理的聚酰亚胺膜和300℃空气氛围下热诱导交联的膜都表现出了典型的增塑现象。但是350℃和400℃的温度下热重排处理之后聚合物膜在压力高达30atm时也未发生塑化现象。通过图8红外分析表明,随着温度达到350℃时,聚合物膜在1557cm
‑1和1016cm
‑1开始出现苯并噁唑环的特征峰,证明了发生热重排反应的聚苯并噁唑气体分离膜表现出优异的抗塑化性能。
[0084]
实施例2
[0085]
单体(b)发生硝化、还原、与6fda发生反应合成聚酰亚胺,反应方程式如图9所示。
[0086]
称取荧光素1.0g,溶于30ml的乙酸中搅拌均匀,再加入1.0g的y(no3)3·
6h2o,反应1h,加入50ml冰冷的去离子水,有大量沉淀析出,搅拌1h,过滤时用大量的冰冷的去离子水冲洗,得到的滤渣于60℃真空烘箱干燥,产率为80%。
[0087]
称取5.0g 2,7'
‑
二硝基荧光素置于100ml三口瓶中,依次加入30ml的thf和20ml的乙醇,搅拌均匀之后加入0.5g的pd/c催化剂,对反应装置抽真空、通入h2,60℃下反应12h,反应结束后,冷却至常温,过滤除去pd/c,趁热不断搅拌缓慢加入适量的去离子水到浊点,冷却结晶,析出固体颗粒,过滤,烘箱60℃下真空干燥。得到黄色粉末2.89g。产率为76.22%。
[0088]
取6fda 1.78g(444.24g/mol,4mmol)加入100ml的三口烧瓶中,加入12ml dmac在低温冰浴和氮气保护下搅拌30min,分两次加入2,7'
‑
二氨基荧光素1.48g(362.34g/mol,4.08mmol),固含量为21wt%,持续搅拌反应14h,形成高粘度的聚酰胺酸溶液,加入5ml的乙酸酐、5ml的无水吡啶,撤掉冰浴。室温下继续反应24h,将粘度很高的聚酰亚胺溶液倒在甲
醇溶液中沉淀,过滤时用甲醇洗涤,除去残留溶剂,80℃烘箱抽真空干燥24h,得到干燥后的聚酰亚胺2.10g。
[0089]
取0.35g干燥好的聚酰亚胺粉末溶解于dmf中,搅拌直至完全溶解,铸膜液固含量32wt%,用0.45μm的ptfe滤膜过滤,将过滤之后的溶液注入平整的表面皿中,然后将其装入自封袋,室温下在平整的台子上放置4天,得到厚度均匀、表面光滑平整的聚酰亚胺致密膜。最后将聚合物膜放在200℃的烘箱中真空干燥24h,除去残余溶剂,最终所得聚合物膜的膜厚在70~80μm之间。
[0090]
将干燥好的聚酰亚胺膜置于碳化炉中,在氮气氛围下,以5℃/min的升温速率分别升温至250℃、275℃和300℃,保温时间为2h,得到在氮气氛围下热诱导交联聚合物膜,分别命名为pi(b)250
‑
n2、pi(b)275
‑
n2、pi(b)300
‑
n2。聚酰亚胺膜在氮气氛围下发生热诱导交联的反应机理图如图10所示。
[0091]
将热诱导交联处理2h之后的致密膜置于碳化炉中,在氮气氛围下,将三个温度下得到的膜以5℃/min的升温速率分别升温至350℃、400℃和450℃,保温时间为1.5h,其中300℃下热诱导交联之后处理得到的pbo膜分别命名为pbo(b)350
‑
6fda、pbo(b)400
‑
6fda、pbo(b)450
‑
6fda。
[0092]
所得热诱导交联的酚酞基聚苯并噁唑气体分离膜中m1、m2、n的值在30至100之间。
[0093]
表2为单体(b)合成的聚酰亚胺膜、300℃氮气氛围下热诱导交联处理2h的膜和pbo(b)350
‑
6fda、pbo(b)400
‑
6fda、pbo(b)450
‑
6fda膜气体透过性和气体选择性。
[0094]
表2
[0095][0096]
从表2中可以看出,由于实施例1和实施例2中聚酰亚胺的结构相似,所以二者的气体渗透性随着温度增加的变化规律相同,由于实施例2聚合物的结构中醚键的存在,导致聚合物链的刚性变差,所以相比于实施例1来说气体渗透性和选择性都稍有降低。同样地,从图11中可以看出,350℃和400℃的温度下发生热重排反应的聚合物膜在压力高达30atm时也未发生塑化现象。
[0097]
以上已经描述了本发明的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1