一种具有高效异相结的氧化钨光催化剂及其制备方法和应用
1.本发明涉及光催化剂技术领域,尤其涉及一种具有高效异相结的氧化钨光催化剂及其制备方法和应用。
背景技术:
2.在众多分解水制备氢能的方法中,光催化分解水被广泛认为是简单、易操作的制氢方法。近些年来,虽然已经公开了一些光催化剂,如中国专利cn109261215a公开了一种光催化分解水制备氢气的催化剂,其公开的石墨烯负载的金属铂催化剂的制氢能力最高为0.8μmol/h,中国专利cn111229260a公开了一种用于可见光下分解水制氢的硫化镉纳米颗粒和二硫化钼纳米带异质结构催化剂及其制备方法,其公开的异质结催化剂ide的制氢能力最高为203.7μmol/(h
·
g),但是可以高效地进行光催化分解水的光催化剂并不多见,主要原因为:催化剂的电子和空穴对分离困难。现有技术一般通过提高催化剂的晶化度、担载助催化剂、进行掺杂改性等方法,使催化剂的电子和空穴对有较好的分离效果,但是这些因素的改善,对光催化活性的提高有限。
3.氧化钨基半导体光催化剂作为光催化剂具有可见光吸收、易制备等优点,但是,氧化钨基半导体光催化剂也由于较高的光生载流子复合几率的问题限制了其在光催化领域的发展。
技术实现要素:
4.本发明的目的在于提供一种具有高效异相结的氧化钨光催化剂及其制备方法和应用,本发明提供的光催化剂具有较高的光催化分解水的活性。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种具有高效异相结的氧化钨光催化剂,包括wo3·
0.33h2o和在所述wo3·
0.33h2o原位相变的m
‑
wo3,
7.所述wo3·
0.33h2o和m
‑
wo3的界面形成异相结。
8.优选的,所述wo3·
0.33h2o和m
‑
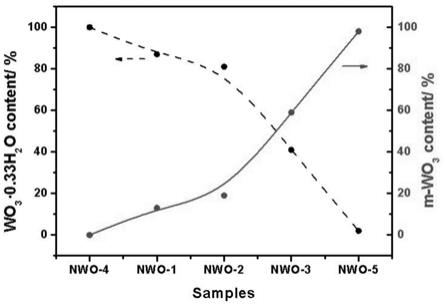
wo3的质量比为(4~9):(1~6)。
9.本发明提供了上述技术方案所述的具有高效异相结的氧化钨光催化剂的制备方法,包括以下步骤:
10.将碱金属钨酸盐与水混合,得到的碱金属钨酸盐水溶液进行第一次水热反应,得到wo3·
0.33h2o;
11.将所述wo3·
0.33h2o与水混合,得到的wo3·
0.33h2o水溶液进行第二次水热反应,得到所述光催化剂。
12.优选的,所述碱金属钨酸盐为钨酸钠或钨酸钾。
13.优选的,所述碱金属钨酸盐水溶液的质量浓度为0.01~0.03g/ml。
14.优选的,所述第一次水热反应和第二次水热反应的ph值独立的为0.5~1.5。
15.优选的,所述第一次水热反应和第二次水热反应的温度独立的为180~200℃。
16.优选的,所述第一次水热反应的时间为3~6h,所述第二次水热反应的时间为1~48h。
17.本发明提供了上述技术方案所述的具有高效异相结的氧化钨光催化剂或上述技术方案所述制备方法得到的具有高效异相结的氧化钨光催化剂在光催化分解水中的应用。
18.优选的,所述光催化剂在光催化分解水体系中的质量浓度为0.001~0.005g/ml。
19.本发明提供了一种具有高效异相结的氧化钨光催化剂,包括wo3·
0.33h2o和在所述wo3·
0.33h2o原位相变的m
‑
wo3,所述wo3·
0.33h2o和m
‑
wo3的界面形成异相结。在本发明中,所述wo3·
0.33h2o为正交相,导带位置为
‑
0.53ev,价带位置为2.67ev,所述m
‑
wo3为单斜相,导带位置为
‑
0.03ev,价带位置为2.77ev,wo3·
0.33h2o和m
‑
wo3的导带和价带的位置适宜,能够形成高效异相结,wo3·
0.33h2o和m
‑
wo3形成的异相结中“结”的构建极大地促进了光生电子和空穴的分离,从而提高了本发明所述光催化剂的光催化活性;同时,由于wo3·
0.33h2o和m
‑
wo3同为wo3体系,使wo3·
0.33h2o和m
‑
wo3更容易满足能级匹配的条件,且两组分近似的电子结构能使光生电子在异相结中更容易实现迁移,显著的提高了wo3·
0.33h2o中光生电子和m
‑
wo3中光生空穴的分离效率,从而提高光催化分解水的活性。根据实施例的结果表明,本发明提供的光催化剂在光催化分解水时,产氢速率为0.3~0.7μmol/h,产氧速率为6.8~7.7μmol/h。
20.本发明还提供了所述光催化剂的制备方法,通过水热反应在wo3·
0.33h2o原位相变生成m
‑
wo3,制备方法简单,易实施。
附图说明
21.图1为实施例3所述光催化剂的xrd图;
22.图2为实施例1~3和对比例1~2所述光催化剂的组分占比图;
23.图3为实施例1~3和对比例1~2所述光催化剂的产氢活性测试图;
24.图4为实施例1~3和对比例1~2所述光催化剂的产氧活性测试图。
具体实施方式
25.本发明提供了一种具有高效异相结的氧化钨光催化剂,包括wo3·
0.33h2o和在所述wo3·
0.33h2o原位相变的m
‑
wo3,所述wo3·
0.33h2o和m
‑
wo3的界面形成异相结。
26.本发明提供的光催化剂包括wo3·
0.33h2o,所述wo3·
0.33h2o导带位置为
‑
0.53ev,价带位置为2.67ev;本发明提供的光催化剂包括m
‑
wo3,所述m
‑
wo3的导带位置为
‑
0.03ev,价带位置为2.77ev;在本发明中,所述wo3·
0.33h2o和m
‑
wo3的界面形成异相结,所述光催化剂的异相结的构建极大地促进了光催化剂的光生电子和空穴的分离,从而提高了光催化剂的光催化活性。
27.本发明对所述光催化剂中wo3·
0.33h2o和m
‑
wo3的质量比没有特殊要求,在本发明中,所述wo3·
0.33h2o和m
‑
wo3的质量比优选为(4~9):(1~6),更优选为(4.5~7):(2~5),最优选为(5~6):(3~4)。
28.本发明提供了上述技术方案所述的具有高效异相结的氧化钨光催化剂的制备方法,包括以下步骤:
29.将碱金属钨酸盐与水混合,得到的碱金属钨酸盐水溶液进行第一次水热反应,得
到wo3·
0.33h2o;
30.将所述wo3·
0.33h2o与水混合,得到的wo3·
0.33h2o水溶液进行第二次水热反应,得到所述光催化剂。
31.在本发明中,如无特殊说明,所用原料均为本领域技术人员熟知的市售产品。
32.本发明将碱金属钨酸盐与水混合,得到的碱金属钨酸盐水溶液进行第一次水热反应,得到wo3·
0.33h2o;在本发明中,所述碱金属钨酸盐优选为钨酸钠或钨酸钾,更优选为钨酸钠,最优选为二水合钨酸钠;所述碱金属钨酸盐水溶液的质量浓度优选为0.01~0.03g/ml,更优选为0.015~0.025g/ml。
33.在本发明中,所述第一次水热反应的ph值优选为0.5~1.5,更优选为0.8~1.2,所述第一次水热反应的ph值优选通过ph调节剂调节得到,所述ph调节剂优选包括强酸,所述强酸优选包括硝酸或盐酸,所述强酸的质量浓度为10~30%。
34.在本发明中,所述第一次水热反应的温度优选为180~200℃,更优选为180℃;所述第一次水热反应的时间优选为3~6h,更优选为4~5h。
35.在本发明中,所述第一次水热反应优选在鼓风干燥箱中进行,本发明对所述第一次水热反应的具体实施方式没有任何特殊的限定,采用本领域技术人员熟知的过程进行即可。
36.在本发明中,所述碱金属钨酸盐通过第一次水热反应首先生成h2wo4,h2wo4继续与水反应形成水和氧化物wo3·
0.33h2o。
37.本发明通过控制第一次水热反应过程中的ph值、水热时间和水热反应温度,得到水和氧化物wo3·
0.33h2o。
38.本发明优选对所述第一次水热反应的固体产物进行后处理,得到所述wo3·
0.33h2o,在本发明中,所述后处理优选包括依次进行:洗涤和干燥,在本发明中,所述洗涤的溶剂优选为乙醇和强酸的混合溶液,所述乙醇和强酸的质量比优选为200ml:100μl,所述强酸优选与上述所述ph调节所用强酸的种类的保护范围相同,在此不在赘述,在本发明中,所述洗涤的次数优选为3~5次,更优选为4次。本发明优选对所述洗涤后的固体产物进行干燥,在本发明中,所述干燥优选为冷冻干燥,所述冷冻干燥的温度优选为
‑
50~
‑
60℃,所述冷冻干燥的时间优选为10~30h,更优选为12~20h。
39.得到wo3·
0.33h2o后,本发明将wo3·
0.33h2o与水混合,得到的wo3·
0.33h2o水溶液进行第二次水热反应,得到所述光催化剂;在本发明中,所述wo3·
0.33h2o水溶液的质量浓度优选为0.012~0.05g/ml,更优选为0.02~0.03g/ml;在本发明中,所述水优选为去离子水。
40.在本发明中,所述第二次水热反应的ph值优选与第一次水热反应的ph值的保护范围相同,在此不再赘述。
41.在本发明中,所述第二次水热反应的温度优选为180~200℃,更优选为200℃;所述第二次水热反应的时间优选为1~18h,更优选为12~24h。
42.在本发明中,所述wo3·
0.33h2o在第二次水热反应时原位相变生成m
‑
wo3,得到所述光催化剂,本发明通过控制第二次水热反应过程中的ph值和水热反应温度,得到m
‑
wo3相。在本发明中,所述光催化剂中m
‑
wo3相的含量与所述第二次水热反应的时间正相关,随着第二次水热反应时间的增长,所述光催化剂中m
‑
wo3相的含量变大。
43.在本发明中,所述第二次水热反应优选在鼓风干燥箱中进行,本发明对所述第二次水热反应的具体实施方式没有任何特殊的限定,采用本领域技术人员熟知的过程进行即可。
44.本发明优选对所述第二次水热反应的固体产物进行后处理,得到所述光催化剂,在本发明中,所述后处理优选与第一次水热反应的后处理的保护范围相同,在此不再赘述。
45.本发明提供的制备方法制备得到的光催化剂形成的异相结中m
‑
wo3是由wo3·
0.33h2o原位相变而成,使wo3·
0.33h2o和m
‑
wo3更容易满足能级匹配的条件,显著的提高了wo3·
0.33h2o中光生电子和m
‑
wo3中光生空穴的分离效率,从而提高光催化分解水的活性。
46.本发明提供了上述技术方案所述的具有高效异相结的氧化钨光催化剂或上述技术方案所述制备方法得到的具有高效异相结的氧化钨光催化剂在光催化分解水中的应用。
47.在本发明中,所述光催化剂在光催化分解水体系中的质量浓度优选为0.001~0.005g/ml,更优选为0.002~0.004g/ml;在本发明中,所述光催化分解水体系的光源优选为疝气灯,所述疝气灯的功率优选为300w;在本发明中,所述光催化分解水体系优选包括光催化分解水制氢体系或光催化分解水制氧体系。
48.在本发明中,所述光催化分解水制氢体系优选包括光催化剂、助催化剂、牺牲剂和水,所述水优选为去离子水;在本发明中,所述助催化剂优选为单质铂或氯铂酸,在本发明中,所述助催化剂的质量和水的体积比优选为(0.3~0.6)g:100ml,在本发明中,所述牺牲剂优选为甲醇,所述牺牲剂和水的体积比优选为(1~2):10。
49.在本发明中,所述光催化分解水制氧体系优选包括光催化剂、牺牲剂和水,所述水优选为去离子水;在本发明中,所述牺牲剂优选为硝酸银,所述牺牲剂的质量浓度优选为0.001~0.002g/ml。
50.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
51.实施例1
52.在搅拌的条件下,将0.9896g二水合钨酸钠和50ml去离子水混合至全部溶解后,滴加质量浓度为20%的hno3,使混合体系的ph值为0.5,在180℃水热反应4小时,用etoh
‑
hno3(体积为200ml:100μl,其中,硝酸体积浓度为0.05%的)混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到wo3·
0.33h2o。
53.将0.62g wo3·
0.33h2o和50ml去离子水混合至全部溶解后,滴加hno3,使混合体系的ph值为1,在200℃下水热反应4小时,用etoh
‑
hno3混合液洗涤4次,在
‑
55℃下冷冻干燥16h,得到所述光催化剂,记为nwo
‑
1,nwo
‑
1中wo3·
0.33h2o和m
‑
wo3的质量比为87:13。
54.实施例2
55.在搅拌的条件下,将0.9896g二水合钨酸钠和50ml去离子水混合至全部溶解后,滴加质量浓度为20%的hno3,使混合体系的ph值为1.5,在180℃水热反应4小时,用etoh
‑
hno3(体积为200ml:100μl,其中,硝酸体积浓度为0.05%的)混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到wo3·
0.33h2o。
56.将0.62g wo3·
0.33h2o和50ml去离子水混合至全部溶解后,滴加hno3,使混合体系
的ph值为1,在200℃水热反应8小时,用etoh
‑
hno3混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到所述光催化剂,记为nwo
‑
2,nwo
‑
2中wo3·
0.33h2o和m
‑
wo3的质量比为81:19。
57.实施例3
58.在搅拌的条件下,将0.9896g二水合钨酸钠和50ml去离子水混合至全部溶解后,滴加质量浓度为20%的hno3,使混合体系的ph值为1,在180℃水热反应4小时,用etoh
‑
hno3(体积为200ml:100μl,其中,硝酸体积浓度为0.05%的)混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到wo3·
0.33h2o。
59.将0.62g wo3·
0.33h2o和50ml去离子水混合至全部溶解后,滴加hno3,使混合体系的ph值为1,在200℃水热反应12小时,用etoh
‑
hno3混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到所述光催化剂,记为nwo
‑
3,nwo
‑
3中wo3·
0.33h2o和m
‑
wo3的质量比为41:59。
60.对比例1
61.在搅拌的条件下,将0.9896g二水合钨酸钠和50ml去离子水混合至全部溶解后,滴加质量浓度为20%的hno3,使混合体系的ph值为0.5,在180℃水热反应4小时,用etoh
‑
hno3(体积为200ml:100μl,其中,硝酸体积浓度为0.05%的)混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到所述光催化剂,记为nwo
‑
4,nwo
‑
4中wo3·
0.33h2o和m
‑
wo3质量比为100:0。
62.对比例2
63.在搅拌的条件下,将0.9896g二水合钨酸钠和50ml去离子水混合至全部溶解后,滴加质量浓度为20%的hno3,使混合体系的ph值为0.5,在180℃水热反应4小时,用etoh
‑
hno3(体积为200ml:100μl,其中,硝酸体积浓度为0.05%的)混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到wo3·
0.33h2o。
64.将0.62g wo3·
0.33h2o和50ml去离子水混合至全部溶解后,滴加hno3,使混合体系的ph值为1,在200℃水热反应48小时,用etoh
‑
hno3混合液洗涤4次,在
‑
55℃冷冻干燥16h,得到所述光催化剂,记为nwo
‑
5,nwo
‑
5中wo3·
0.33h2o和m
‑
wo3的质量比为0:100。
65.测试例1
66.将实施例3所述的光催化剂进行xrd测试,测试结果如图1所示。衍射角为14
°
、18
°
、27
°
、36
°
和49
°
处出现的峰为wo3·
0.33h2o的特征峰,与wo3·
0.33h2o标准卡片(pdf#72
‑
0199)相一致;衍射角为23
°
、34
°
、39
°
和48
°
处出现的峰为m
‑
wo3特征峰,与m
‑
wo3标准卡片(pdf#71
‑
2141)相一致,表明实施例3成功制备了wo3·
0.33h2o和m
‑
wo3的异相结光催化剂。所述wo3·
0.33h2o导带位置为
‑
0.53ev,价带位置为2.67ev;本发明提供的光催化剂包括m
‑
wo3,所述m
‑
wo3的导带位置为
‑
0.03ev,价带位置为2.77ev。
67.应用例1
68.将实施例1~3和对比例1~2制备得到的光催化剂进行催化活性测试:
69.产氢活性测试:将0.1g光催化剂和90ml去离子水置于反应器中,在搅拌的条件下混合构成悬浮体系,依次加入50μl氯铂酸(浓度10mg/ml,以pt计)和10ml甲醇。将反应器接入光催化测试系统,抽真空20min去除水中含有的溶解氧,开氙气灯进行照射,在300w的氙灯光源的照射下进行光催化分解水反应。开灯后前2个小时先进行光沉积反应,光沉积结束后移开光源,抽真空5min去除光催化测试循环系统中光沉积反应生成的气体,然后移回光源进行光催化分解水反应,并从此时开始计时,每1h取样1次,取样至开始计时后第4h。产氢活性由与光催化测试系统相接连的气相色谱仪进行展示。
70.产氧活性测试:将0.1g光催化剂和100ml去离子水置于反应器中,在搅拌的条件下混合构成悬浮体系,加入0.1699g硝酸银。将反应器接入光催化测试系统,抽真空20min去除水中含有的溶解氧,开氙灯进行照射,在300w的氙灯光源的照射下进行光催化分解水反应。开灯后开始计时,每1h取样1次,取样至开始计时后第4h。产氧活性由与光催化测试系统相接连的气相色谱仪进行展示。
71.测试结果如图3所示,由图3得出的数据现列于表1。由图3和表1得出,本发明提供的光催化剂由于形成了wo3·
0.33h2o和m
‑
wo3的界面异相结,极大地促进了催化剂光生电子和空穴的分离,从而提高了光催化剂的光催化活性,在光催化分解水时,产氢速率为0.3~0.7μmol/h,产氧速率为6.8~7.7μmol/h,而对比例1制备的催化剂仅为wo3·
0.33h2o,产氢速率为0.02μmol/h,产氧速率为3.7μmol/h,对比例2制备的催化剂仅为m
‑
wo3,产氢速率为0μmol/h,产氧速率为6.2μmol/h,均低于实施例1~3得到的催化剂产品。
72.表1不同光催化剂产氢产氧活性
73.序号h2生成速率μmol/ho2生成速率μmol/hnwo
‑
10.76.8nwo
‑
20.57.7nwo
‑
30.37.5nwo
‑
40.023.7nwo
‑
506.2
74.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1