一锅连投合成奥氮平中间体的方法及专用固体碱催化剂与流程
1.本发明涉及一种奥氮平中间体——2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩的合成方法,属于医药合成技术领域。
背景技术:
2.奥氮平(olanzapine),化学名为:2
‑
甲基
‑4‑
(4
‑
甲基
‑1‑
哌嗪基)
‑
10h
‑
噻酚并[2,3
‑
b][1,5]苯二氮杂,是美国eily公司研发的精神分裂症治疗药物。1996年10月在美国首次上市,我国于1998年批准进口。本品系新型非典型抗精神病药,作用于中枢神经系统,具有5
‑
ht2和多巴胺d2双重拮抗作用,与氟哌啶醇(haloperidol)等抗精神病药物相比,临床疗效好,锥体外不良反应小,临床应用广泛。
[0003][0004]2‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩作为奥氮平的药物中间体,其合成方法优劣对于提高药物合成产品质量,减少副产物含量具有重要经济意义。
[0005]2‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩的合成,目前使用较多的是cn102225941a、cn102250116a等公开的合成路线,即以dmf(或thf)为溶剂,以硫、丙醛和丙二腈为原料,三乙胺作催化剂,关环得5
‑
甲基
‑2‑
氨基
‑3‑
氰基噻吩,再与邻硝基氟苯在氢化钠作用下生成5
‑
甲基
‑2‑
(2
‑
硝基苯氨基)
‑3‑
氰基噻吩,合成路线如下所示。
[0006][0007]
该方法将两步缩合反应分开进行,收率偏低,并且体系溶剂无法回收或者耗能很大,同时母液损失较大。
技术实现要素:
[0008]
针对上述问题,本发明提供了一锅连投合成奥氮平中间体的方法及专用固体碱催化剂。本发明首先制备了一种活性、稳定性及抗磨损性能好的新型负载型固体碱催化剂,以硫粉、丙醛、丙三腈为原料,以丙酮作为溶剂,在新型负载型固体碱催化剂的存在下,加入相转移催化剂四丁基溴化铵缩合得到2
‑
氨基
‑5‑
甲基噻吩
‑3‑
睛,将上步反应料液直接作为底物,采用滴加2
‑
氟硝基苯的丙酮溶液方式加料,再经缩合反应得到2
‑
(邻硝基苯氨基)3
‑
氰基
‑5‑
甲噻吩,产品收率达88
‑
90%。
[0009]
本发明第一个目的是提供合成奥氮平中间体专用的固体碱催化剂的制备方法,其特征是,将蒙脱石或活性炭浸渍到ph=4
‑
5的酸性溶液(冰醋酸和醋酸钠调配)中,然后再加入镁盐和铝盐,混合均匀后将溶液调制ph=9
‑
10,搅拌反应;反应完毕后经过滤、滤饼焙烧、研磨得到固体碱催化剂。
[0010]
所述镁盐为氯化镁或硝酸镁;铝盐为氯化铝或者硝酸铝。
[0011]
所述的焙烧、研磨为:500
‑
600℃下焙烧2
‑
3h,冷却,研磨至100
‑
200目的颗粒产品。
[0012]
进一步的,蒙脱石或活性炭在ph=4
‑
5的酸性溶液中浸渍2
‑
5h;将溶液调制ph9
‑
10后搅拌反应3
‑
8h。
[0013]
所述各物料之间的质量比为,蒙脱石(活性炭):氯化铝(硝酸铝):氯化镁(硝酸镁)=1:0.5
‑
0.8:0.6
‑
1.0,优选为1:0.6:0.8。
[0014]
本发明使用的原料蒙脱石(活性炭)首先粉碎、过筛,粒径在0.2
‑
1mm。
[0015]
本发明第二个目的是提供了一锅连投合成奥氮平中间体的方法,包括以下步骤:
[0016]
(1)制备5
‑
甲基
‑2‑
氨基
‑3‑
氰基噻吩
[0017]
将硫粉、相转移催化剂四丁基溴化铵以及上述方法制备的固体碱催化剂加入到丙酮溶剂中,滴加丙醛,搅拌反应,然后滴加丙二腈的丙酮溶液,滴毕,15℃以下进行缩合反应得到含5
‑
甲基
‑2‑
氨基
‑3‑
氰基噻吩的反应液;
[0018]
(2)制备2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩
[0019]
上述反应液补加固体碱催化剂,然后滴加2
‑
氟硝基苯的丙酮溶液,滴加完毕后15℃以下进行缩合反应;
[0020]
反应完毕后减蒸掉部分丙酮,抽滤出固体碱催化剂;然后滤液加水,降温析晶;再经水洗、干燥得到2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩。
[0021]
具体的合成路线如下所示:
[0022][0023]
进一步的,步骤(2)抽滤得到的固体碱催化剂,加水浸泡后,放入马弗炉500
‑
600℃下焙烧3
‑
4h,冷却,研磨至100
‑
200目的颗粒产品,继续进行套用。
[0024]
其中,步骤(1)丙醛滴加完毕后搅拌反应0.5
‑
2h;丙二腈滴加完毕后保温反应1
‑
2h;步骤(2)滴加完毕后保温反应2
‑
3h。
[0025]
所述步骤(1)丙二腈、硫粉和丙醛的摩尔比为1:1.0~1.5:1.2~1.8;四丁基溴化铵加入量为硫粉质量的0.5~3.0%,优选1%。
[0026]
以硫粉的质量计,步骤(1)加入固体碱催化剂的量为2~5%,优选3%。步骤(2)加入固体碱催化剂的量为5~8%,优选为6%。
[0027]
所述步骤(2)使用的2
‑
氟硝基苯与丙二腈的摩尔比为1.2~1.8:1。
[0028]
本发明的原理是:
[0029]
1、采用浸渍方法制备新型负载型固体碱催化剂,具备较多的碱性活性位点(碱性高),有利于加快硫粉与丙醛生成硫醇,提高反应速率及选择性,同时该催化剂有较高的热稳定性,有利于催化剂的回收重复利用;
[0030]
2、步骤(1)中使用均相相转移催化剂四丁基溴化铵有利于固体碱催化剂与原料接触,有利于原料降量及大幅度提高反应速率;
[0031]
3、步骤(1)中以丙酮替代传统溶剂dmf体系,既使得两步合成有可能改成一锅投料(dmf在较强碱体系下容易发生分解),也增加了丙酮的回收套用(dmf回收较难,耗能太大),减少母液损失,提高了收率;同时减蒸丙酮后,可以大幅度减少析晶的水量,故大幅度减少废水量。
[0032]
与之前文献报道的工艺相比,本发明具有以下优势:
[0033]
1、本发明制备的固体碱催化剂的比表面积和孔体积较大,含有丰富的中大孔结构,活性及稳定性高,具有良好的抗磨损性能,催化剂便于分离回收,可多次重复套用。
[0034]
2、一锅连投,改变dmf体系以及强碱体系,同时可以对丙酮进行回收套用,并减少母液损失。
[0035]
3、该反应工艺条件合理,缩短反应时间,产品收率可达88
‑
90%,具有广阔的经济效益和生产前景。
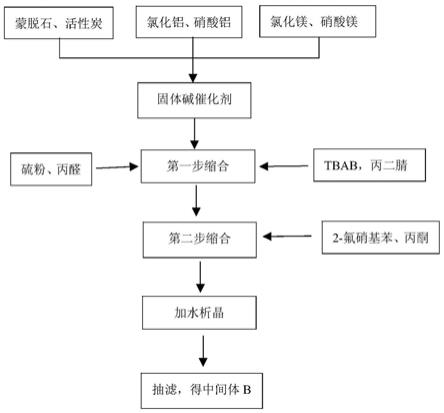
附图说明
[0036]
图1为本发明采用固体碱催化剂一锅连投合成奥氮平中间体的工艺流程图。
具体实施方式
[0037]
以下结合实施例对本发明进一步说明。其工艺流程图如图1所示。本实施例使用的原料蒙脱石(活性炭)首先粉碎、过筛,粒径在0.2
‑
1mm。
[0038]
本实施例1和实施例3使用的固体碱催化剂的制备方法:在室温条件下,将100g蒙脱石加入到ph=4
‑
5的酸性溶液(3.5
‑
3.9g冰醋酸和2.8
‑
3.0g醋酸钠加入适量的水充分溶解后,再定容至1l)中,室温下浸渍3
‑
4h;将60g氯化铝和80g氯化镁加入上述酸性溶液中,混合均匀后将溶液调制ph=9
‑
10(使用15
‑
25%氨水调节ph),搅拌反应5
‑
6h,过滤,滤饼在500
‑
600℃下焙烧2
‑
3h,冷却,研磨至100
‑
200目的颗粒产品,待用。
[0039]
本实施例2使用实施例1步骤(3)回收的固体碱催化剂。
[0040]
实施例1:
[0041]
(1)制备5
‑
甲基
‑2‑
氨基
‑3‑
氰基噻吩
[0042]
称取20g的硫粉、0.2g四丁基溴化铵以及0.6g固体碱催化剂加入到80g丙酮溶剂
中,在10
‑
15℃条件下,将45.6g丙醛滴加至上述溶液中,滴加1
‑
2h,搅拌反应1h,将36g丙二腈溶解在36g丙酮中,溶清后开始滴加丙二腈的丙酮溶液,滴加过程中控制温度15℃以下,滴加完毕后保温反应1
‑
2h。保温结束后进行tlc检测,若反应液浅于对照点,反应结束;若不浅于,继续反应直至浅于对照点。
[0043]
(2)制备2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩
[0044]
第一步缩合反应完毕后,补加1.2g固体碱催化剂,称取112g 2
‑
氟硝基苯与60g丙酮混匀,然后滴加2
‑
氟硝基苯的丙酮溶液,滴加过程中控制温度在15℃以下,滴加完毕后保温2
‑
3h。
[0045]
第二步缩合反应完毕后减蒸130g丙酮,抽滤,用适量的丙酮淋洗滤饼(固体碱催化剂),滤饼回收待用;滤液和洗涤液合并后(共80g)加入1600g纯化水,室温搅拌0.5h,再降温至
‑
10
‑
0℃,析晶1.5h。抽滤,再用60g的纯化水洗涤滤饼;将滤饼放入烘箱,50
‑
60℃真空干燥6
‑
8h,得到2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩固体126.9g,纯度99.2%,产品总收率89.6%。
[0046]
(3)固体碱催化剂收回套用
[0047]
步骤(2)抽滤得到的固体碱催化剂,加水浸泡后,放入马弗炉500
‑
600℃下焙烧3
‑
4h,冷却,研磨至100
‑
200目的颗粒产品,继续进行套用。
[0048]
实施例2:
[0049]
(1)制备5
‑
甲基
‑2‑
氨基
‑3‑
氰基噻吩
[0050]
称取20g的硫粉、0.2g四丁基溴化铵以及0.6g固体碱催化剂(实施例1回收的固体碱催化剂)加入到90g丙酮溶剂中,在10
‑
15℃条件下,将46.0g丙醛滴加至上述溶液中。滴加1
‑
2h,搅拌反应1h,将40g丙二腈溶解在40g丙酮中,溶清后开始滴加丙二腈的丙酮溶液,滴加过程中控制温度15℃以下,滴加完毕后保温1
‑
2h。
[0051]
(2)制备2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩
[0052]
第一步缩合反应完毕后,补加1.2g固体碱催化剂,称取112g 2
‑
氟硝基苯与70g丙酮混匀,然后滴加2
‑
氟硝基苯的丙酮溶液,滴加过程中控制温度在15℃以下,滴加完毕后保温2
‑
3h。
[0053]
第二步缩合反应完毕后减蒸130g丙酮,抽滤,用适量的丙酮淋洗滤饼(固体碱催化剂),滤饼回收待用;滤液和洗涤液合并后(共100g),然后加入1700g纯化水,室温搅拌0.5h,再降温至
‑
10
‑
0℃,析晶1
‑
1.5h。抽滤,再用60g的纯化水洗涤滤饼;将滤饼放入烘箱,50
‑
60℃真空干燥6
‑
8h,得到2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩固体约125.8g,纯度99.0%,产品总收率达88.82%。
[0054]
(3)固体碱催化剂收回套用
[0055]
步骤(2)抽滤得到的固体碱催化剂,加水浸泡后,放入马弗炉500
‑
600℃下焙烧3
‑
4h,冷却,研磨至100
‑
200目的颗粒产品,继续进行套用。
[0056]
实施例3:
[0057]
(1)制备5
‑
甲基
‑2‑
氨基
‑3‑
氰基噻吩
[0058]
称取20g的硫粉、0.3g四丁基溴化铵以及0.6g固体碱催化剂加入到100g丙酮溶剂中,在10
‑
15℃条件下,将45.0g丙醛滴加至上述溶液中。滴加1
‑
2h,搅拌反应1h,将36g丙二腈溶解在50g丙酮中,溶清后开始滴加丙二腈的丙酮溶液,滴加过程中控制温度15℃以下,
滴加完毕后保温1
‑
2h。
[0059]
(2)制备2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩
[0060]
第一步缩合反应完毕后,补加1.2g固体碱催化剂,称取113g 2
‑
氟硝基苯与80g丙酮混匀,然后滴加2
‑
氟硝基苯的丙酮溶液,滴加过程中控制温度在15℃以下,滴加完毕后保温2
‑
3h。
[0061]
第二步缩合反应完毕后减蒸150g丙酮,抽滤,用适量的丙酮淋洗滤饼(固体碱催化剂),滤饼回收待用;滤液和洗涤液合并后(共120g),然后加入1800g纯化水,室温搅拌0.5h,再降温至
‑
10
‑
0℃,析晶1
‑
1.5h。抽滤,再用60g的纯化水洗涤滤饼;将滤饼放入烘箱,50
‑
60℃真空干燥8h,得到2
‑
(邻硝基苯氨基)
‑3‑
氰基
‑5‑
甲基噻吩固体为127.1g,纯度99.3%,产品总收率达89.74%。
[0062]
(3)固体碱催化剂收回套用
[0063]
步骤(2)抽滤得到的固体碱催化剂,加水浸泡后,放入马弗炉500
‑
600℃下焙烧3
‑
4h,冷却,研磨至100
‑
200目的颗粒产品,继续进行套用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1