色谱材料及生产其的方法与流程
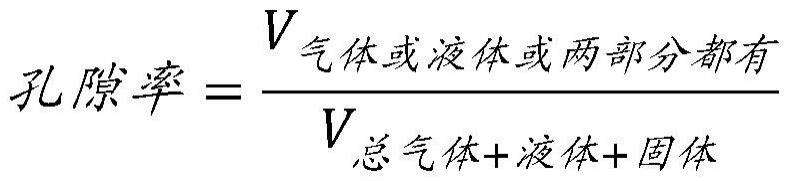
色谱材料及生产其的方法
1.本发明涉及用于在液体介质中吸附材料分离的包括基于聚合物网络材料的自支撑双连续分离基质的色谱材料,以及生产包括基于聚合物网络材料的自支撑双连续分离基质的色谱材料的方法。
2.色谱是一种通用的分离技术,其使用目的分子在固定相和流动相之间的分布来进行分子分离。固定相是指多孔介质和吸收的固定溶剂。具有相关联的端盖、配件和管的柱是最常见的配置,其中介质填充到管或柱中。将流动相泵送通过柱。在柱的一端引入样品,并且各种组分(目标物质和污染物)与固定相相互作用,并以不同的速度吸附到介质上或介质中或穿过柱。在柱的另一端收集或检测分离的组分。通过将洗脱剂溶剂泵送通过柱,可以在单独步骤中释放吸附的组分。除了其他方法之外,色谱方法还包括凝胶色谱、离子交换色谱、疏水相互作用色谱、反相色谱、亲和色谱、免疫吸附色谱、凝集素亲和色谱、离子亲和色谱和其它这类熟知的色谱方法。
3.已知多糖凝胶在制备用于分离生物分子混合物的材料中起重要作用。在使这些凝胶特别令人关注的特征中,可以提及它们与蛋白质和其它生物分子接触的惰性以及它们的多孔结构。另一个重要的性质是它们对碱性条件的抗性,这在需要频繁再生/灭菌凝胶的大规模分离过程中是非常重要的。
4.多糖凝胶用于各种类型的色谱。一个这样的实例是凝胶过滤,由此对样品成分进行尺寸分级。这是其中惰性是至关重要的应用,因为凝胶材料本身和样品分子之间的任何相互作用使得分离效率低并且甚至可能完全破坏结果。商业上重要的色谱技术的另一个实例是离子交换色谱。用于离子交换色谱的几种材料衍生自右旋糖酐、琼脂糖或纤维素。选择多糖凝胶作为载体基质是基于它们的惰性和用于引入离子交换基团的得到确认的衍生化学。另一个实例是亲和色谱。同样,多糖凝胶在许多应用中是优选的,这是由于可用于引入亲和配体的便利技术的数量以及生物分子的非特异性结合的最低限度。
5.多糖凝胶可以制备成各种形状,例如或多或少规则的珠粒,如球体,也可以是膜的形状等,并且可以用作制备色谱介质的基础基质,以及用作通常用于各种生物分子如细胞、酶、抗体等的载体基质。
6.多糖凝胶通常具有小直径扩散孔,分子尺寸为约10至500nm,其可以仅由在流动相中的组分(目标物质和/或污染物)经由扩散达到。为了将结合组分有效地输送到凝胶,对流相包围多糖凝胶对流相,在对流相和扩散凝胶相之间形成界面。多糖凝胶最广泛地使用于直径约50至100μm的颗粒形状。将这些颗粒填充到色谱柱中,在色谱柱中,颗粒之间的间隙相形成对流相,从而使分子快速地运输通过色谱柱并到达珠粒的表面,即到达多糖凝胶的表面。除了形成通过将珠粒填充到柱中而与扩散相交界的对流相之外,对流相还可以由大直径对流孔(也称为“流过孔”或“大孔”)形成,所述大直径对流孔可以提供通过凝胶的流动通路或通道。固化的多糖凝胶在大直径对流孔周围形成所谓的“桥”。小直径扩散孔位于由固化多糖形成的所述桥内。当这种多孔多糖凝胶被填充在色谱柱中并且通过柱施加液体流时,该流将主要经由大直径对流孔通过凝胶。因此,待分离的组分(目标物质和/或污染物)也通过对流流输送到凝胶的内部,这是比扩散快得多的输送方式。因此,在这种多孔多糖凝
胶中只有短距离需通过扩散来覆盖。图1显示了多糖颗粒和多糖膜中的对流和扩散流动路径的比较。
7.从例如us 4,935,365a中已知可生产含有大孔的多糖。其中所述的凝胶由于大孔而具有相当大的表面积,这一事实增加了其在一些应用如细胞培养以及某些色谱技术中的用途。然而,us 4,935,365a中的大孔不是相互连接的,因此没有经由这些孔的通流。
8.孔的直径越小,“桥”/扩散路径长度(它是桥直径的一半长度)也越小。大直径对流孔和桥之间的相关性通过几何考虑可得到。特别地,在恒定的孔体积分数(对流孔隙率)的情况下,大直径对流孔变得越大,桥必须变得越大,因为桥仅代表大直径对流孔之间的空间。如果存在较少的大直径对流孔(具有恒定体积分数的较大孔也意味着较少的孔),则它们之间的空间必须增加。因此,在多孔多糖材料具有相当小的大直径对流孔的情况下,桥内的扩散路径长度也相当小,并且在非常短的扩散时间内可进入多孔桥内的扩散孔。这种几何上给定的相关性强烈地表明为什么较小的孔径对于这种材料内的色谱操作是有利的。
9.在生产具有两种类型的孔(即小直径扩散孔和大直径对流孔)的多孔多糖材料中,对流孔的孔径(以及桥尺寸)和多糖材料的结构形态受到以下相关工艺参数的影响:
10.·
表面活性剂的体积比(vol%)
11.·
表面活性剂的类型
12.·
有机溶剂(与水不混溶的有机相)的体积比(vol%)
13.·
含水胶凝相的体积
14.·
有机溶剂的类型(单一溶剂或溶剂的混合物)
15.·
含水胶凝相中多糖的浓度
16.·
在多糖的含水溶液与基本上与水不混溶的有机相混合期间的搅拌速度。
17.us 5,723,601a描述了具有两种类型的孔的多孔多糖材料,即小直径扩散孔和大直径对流孔。在us 5,723,601a的实施例中,生产多孔多糖材料,其中对流孔具有10μm的最小直径。尽管us 5,723,601a也提及了小至0.5μm的对流孔径,但从技术观点来看,不可能通过us 5,723,601a教导的方法生产具有直径小于10μm的大直径对流孔的多孔多糖材料,因为us 5,723,601a没有描述多糖浓度、表面活性剂(类型和浓度)和有机溶剂的合适组合以生产具有这种小对流孔的多孔多糖材料。特别地,us 5,723,601a教导了在较高的搅拌器速度下使用较小的表面活性剂浓度的情况下可以获得较小的对流孔。然而,当在最高可能的搅拌器速度和在us 5,723,601a中描述的实验条件下降低表面活性剂浓度时,对于互连的对流孔可获得的最小对流孔径为约6.5μm。当在us 5,723,601a中描述的实验条件下进一步降低表面活性剂浓度时,所获得的孔结构不是互连的孔结构,而是闭孔泡沫结构,这将在下文说明。泡沫由部分或多或少连接的球状结构组成,作为扩散相之间的对流相(大孔),即大孔不互连。固有地较差连接性导致在相当的孔径(即球状结构的直径)下具有非常低的渗透率的结构,因为没有经由这些孔的通流。因此,泡沫结构只能以非常不均匀和不适当的方式穿过,这导致流体在色谱介质中均匀分布的缺点,以及除了较低的渗透率之外,还导致动态结合的缺点。
18.us 7,479,233b2描述了用于吸附或基于色谱的分离的多孔涂覆介质,其包括基底基材(base substrate),所述基底基材是多孔无纺织物,自支撑结构,并且在基材的所有表面的至少一部分上由多糖形成的一个或多个多孔涂层。在涂覆介质的生产方法中,首先用
多糖层涂覆惰性基材,然后使涂覆的基材经受胶凝剂使得多糖形成多孔水凝胶涂层。由于这种特定的生产方法(涂覆和随后凝胶化),惰性基材构成多孔水凝胶涂层的结构,即水凝胶涂层的孔结构遵循惰性基材的孔结构。由于需要存在大量的惰性基材作为多孔涂层的支撑(其用作结合目标化合物和/或污染物的色谱介质),多孔结合相的可能量受到大量限制,并且吸附介质的可能结合能力受到负面影响。
19.us 8,298,657b2描述了一种功能性的多孔互穿聚合物网络(ipn),其包括成孔(porogenic)支撑织物形式的第一聚合物网络和在第一聚合物网络存在下合成、凝胶化和/或交联的第二聚合物网络,所述成孔支撑织物由包括非织造纤维的预成形网络形式的线性聚合物组成。在ipn的生产中,第一聚合物网络的纤维的一部分被去除以形成形状基本上为圆柱形的孔,因为去除的纤维具有约1.0毫米的最小长度。与us 7,479,223b2中一样,羊毛纤维的强制性存在也限制了结合聚合物的最大可能量,并且对结合能力具有负面影响。另外,使用非织造纤维来溶解它们作为形成圆柱形孔的方法导致显著更高的弯曲度,如本领域技术人员所知,这导致在相当的孔径直径下较低的渗透率。
20.us 2011/0120947a1描述了用于色谱过程的交联的纤维素水合物膜,所述交联的纤维素水合物膜具有由“微孔”和“超微孔”组成的多孔双重结构。us2011/0120947a1的膜必须由醋酸纤维素组成,其在制造过程中通过这种材料的膨胀和皂化导致一种具有对流和扩散组分的材料。然而,由再生纤维素组成的这些材料不是自胶凝材料或由自胶凝材料构成的相,并且存在于这些材料中的扩散孔非常小并且难以控制。
21.us 7,316,919b2描述了多孔载体和非自支撑交联凝胶的复合材料,所述非自支撑交联凝胶衍生自自由基聚合反应,所述自由基聚合反应含有10-3000nm的“大孔”并且位于载体结构的孔内。然而,这些材料在其最大凝胶孔隙率和对流孔隙率方面是有限的。另外,由于合成聚合物凝胶的性质,复合材料中的孔隙率和孔径分布都难以控制。与上述us 7,479,223b2和us 8,298,657b2一样,载体材料的存在显著减少了结合聚合物的数量,从而大大损害了这种复合材料的渗透率和可能结合能力。
22.鉴于上述情况,本发明的潜在技术问题是提供色谱材料,利用该色谱材料可以更快速和更有效地进行色谱过程;以及提供生产所述色谱材料的方法。
23.上述技术问题的解决方案是通过提供权利要求中表征的实施方案来实现的。
24.特别地,提供了包括基于聚合物网络材料的自支撑双连续分离基质的色谱材料,所述基质包含第一连续相和第二连续相,所述色谱材料任选地包括第三惰性相,其中所述第一连续相是由自凝胶多糖形成的所述基质的一部分,并且其中所述第二连续相是所述基质的一部分,其限定在所述第一连续相之间的非圆柱形大直径连续连接的对流孔,其中所述大直径连续连接的对流孔具有0.1μm至6.0μm的中值孔径,并且其中所述第三惰性相不构成所述第二连续相的结构。
25.本发明的色谱材料包括作为自支撑分离基质的分离基质。如本文所用,表述“自支撑分离基质”意指由于其连续连接的凝胶结构,分离基质固有地尺寸稳定,并且可以在不需要惰性基材作为支撑基材的情况下产生和处理。这不排除在需要时引入第三惰性相作为增强基材以进一步增强机械强度或提高处理或整合到色谱装置中的容易性的可能性。
26.本发明的色谱材料的自支撑基质具有双连续结构。如本文所用,表述“双连续的(bi-continuous)”或“双连续的(bicontinuous)”意指分离基质具有包括两个体积大的(大
部分)连续相的结构,所述两个体积大的(大部分)连续相彼此渗透并且各自(大部分)本身是连续的(互连的)。因此,(大部分)连续相中至少90%,优选地至少95%,并且更优选地至少98%的体积元素可以从该体积大的相中的任何点到达,而不离开该相。根据本发明的特定优选实施方案,连续相内的每个体积元素可以从该体积大的相内的任何点到达,而不离开该相。这可以在分离基质的3d结构中通过目视检查至少64*103·
μm3的体积元素来看到。如何通过共焦激光扫描显微镜获得3d结构是本技术领域的技术人员已知的,并且其实施例可以在ley等人的科学出版物,journal of membrane science,第564卷,2018年,第543-551页(第545页)中找到。
27.本发明的色谱材料的分离基质的第一连续相是由自胶凝多糖形成的基质的一部分,即第一连续相包括至少90%,优选地至少95%,更优选地至少98%,并且更优选地至少99%的自胶凝多糖。根据特别优选的实施方案,第一连续相包括100%的自胶凝多糖,即第一连续相由自胶凝多糖组成。第一连续相通常具有约10至500nm的分子尺寸的小直径扩散孔。第一相(大部分)是连续连接的,即第一相内的至少90%,优选地至少95%,并且更优选地至少98%的体积元素可以从第一相内的任何点到达,而不离开第一相。根据本发明的特定优选实施方案,第一相内的每个体积元素可以从第一相内的任何点到达,而不离开第一相。
28.如本文所用,“胶凝”是其中聚合物溶液,特别是多糖溶液固化并因此形成聚合物凝胶的过程,其中溶剂(例如水或甘油)填充聚合物凝胶内的多孔空间。溶剂可以改善凝胶的(储存)稳定性。对于自胶凝(self-gelling)/自胶凝(self-gelled)聚合物/多糖凝胶,不必涉及外部或另外的聚合物或单体或交联剂来形成导致聚合物/多糖凝胶形成的物理或共价键。
29.自胶凝多糖(具有小直径扩散孔)是多孔的,即多糖具有至少20%的孔隙率。自胶凝多糖内的孔是多糖凝胶典型的小直径扩散孔。本文所用的孔隙率被定义为由气体或液体填充的材料的体积(v
气体或液体或两部分都有
)除以包括固体体积在内的材料的总体积(v
总气体+液体+固体
)。
[0030][0031]
根据本发明的优选实施方案,自胶凝/自胶凝多糖为选自以下的一种或多种:琼脂糖、琼脂、琼脂胶、κ-卡拉胶、ι-卡拉胶、λ-卡拉胶、结冷胶、直链淀粉、凝胶多糖、海藻酸盐和鼠李聚糖胶(rhamsan gum)。通过在制备期间使用单一乳液,这些自胶凝/自胶凝多糖有利地允许形成具有小孔和大孔的分离基质,即具有小直径扩散孔和大直径对流孔的结构(双孔结构)。此外,所述亲水性多糖表现出低的非特异性结合。根据本发明特别优选的实施方案,所述自胶凝/自胶凝多孔多糖是琼脂糖。
[0032]
本发明的色谱材料的分离基质的第二连续相是基质的一部分,其限定了在第一连续相之间的非圆柱形大直径连续连接的对流孔。非圆柱形大直径对流孔(大部分)是连续连接的(互连的),即,通过仅移动通过非圆柱形大直径对流孔,至少90%,优选地至少95%,并且更优选地至少98%的非圆柱形大直径对流孔的孔可以从非圆柱形大直径对流孔的另一个孔的任何点到达。根据本发明的特别优选的实施方案,通过仅移动通过非圆柱形大直径对流孔,可以从非圆柱形大直径对流孔的另一个孔的任何点到达非圆柱形大直径对流孔的
任何孔。非圆柱形大直径对流孔位于第一连续相(自胶凝多糖)之间,即自胶凝多糖形成围绕非圆柱形大直径对流孔的多孔凝胶桥。所述大直径对流孔是非圆柱形的,即所述孔基本上是球形的,其中基本上球形的孔的部分通常重叠以形成连续的对流孔结构。
[0033]
根据本发明,非圆柱形大直径连续连接的对流孔具有0.1μm至6.0μm的中值孔径。迄今为止,本领域尚未描述包括基于聚合物网络材料的自支撑双连续分离基质而没有惰性相作为构成分离基质的第二连续(孔)相的支撑材料的色谱材料,所述色谱材料具有如此小的非圆柱形连续连接的大直径对流孔,因为没有已知的用于制备此类色谱材料的合适方法。中值孔径可以例如确定为来自ley等人的科学出版物,journal of membrane science,第564卷,2018年,第543-551页(第546-548页)中的方法的所有孔数据的中值。优选地,中值孔径为0.5μm至5.0μm,最优选地为1.0μm至3.0μm。由于非圆柱形连续连接的大直径对流孔具有在上述范围内的中值孔径,因此扩散路径长度也很小(参见图4)并且在很短的扩散时间内就可以到达多孔桥内的小直径扩散孔,有利地允许非常快速且有效地操作色谱过程。
[0034]
尽管本发明的色谱材料的分离基质是自支撑的固有稳定的分离基质,但本发明的色谱材料也可任选地包括第三惰性相作为增强材料,以进一步增强色谱材料抵抗外部应力,例如压力或破坏辐射。在第三惰性相作为增强材料存在的情况下,第三惰性相不构成分离基质的第二连续相的结构。也就是说,第三惰性相独立于第二连续相的结构随机地分布在基于聚合物网络材料的自支撑双连续分离基质中,并且第二连续相的孔结构不遵循惰性相的结构,如us7,479,233b2中所述的色谱材料。
[0035]
作为本发明的色谱材料中的分离基质的另外增强材料的第三惰性相由于增加分离基质的稳定性可以促进色谱材料的生产和使用。用作增强材料的第三惰性相没有特别限制,并且可以例如是羊毛材料或非织造材料(干法成网、湿法成网、纺法成网、熔喷法)、织造材料、织物、网状织物、开孔泡沫、网状泡沫或其它可渗透的多孔材料。商业上可获得的增强材料是例如来自freudenberg的非织造novatexx 2465;来自berry非织造reemay 2016以及网状织物sefar 07-200/35。
[0036]
根据本发明的优选实施方案,另外地包括第三惰性相的色谱材料具有第三惰性相的体积与包括第三惰性相的色谱材料的总体积的比值为0.03至0.60,更优选为0.05至0.40,最优选为0.08至0.25。如本文所用,“第三惰性相的体积”(v)是第三惰性相所占的体积。通过知道下面材料的密度可以由其质量(m)计算第三惰性相的体积(v):
[0037][0038]
如本文所用,“包括第三惰性相的色谱材料的总体积”是色谱材料的物理尺寸的总体积,其包括第三惰性相体积和两个连续相的体积。所述总体积可以通过测量色谱材料的外部尺寸如直径和厚度来测定,这取决于其宏观外观。
[0039]
如果上述体积比值低于上述范围,则可降低用作增强材料的第三惰性相的稳定性增强效果。如果上述体积比值高于上述范围,则色谱材料的结合能力和渗透率将降低。
[0040]
用作增强材料的第三惰性相优选地具有一种或多种以下性质:
[0041]
·
碱稳定性,即增强材料耐受0.1n naoh至少5h,更优选地至少24h,而无明显降解或功能丧失;
[0042]
·
γ稳定性,即增强材料耐受至少10kgy,更优选地25kgy,最优选地50kgy,而无明显降解或功能丧失;
[0043]
·
拉伸强度:》5.2m pa,优选》6.8mpa(拉伸试验使用2.5kn
[0044]
试验机(zwick&roll)进行。测量期间的温度为20℃/室温。拉伸样本为150mm长,20mm宽。将每个样本放置在试验机的样本架中心。所有测量的拉伸速度为50mm/min。拉伸强度值被定义为
[0045]
样品断裂时的最高拉伸强度(rm)。);
[0046]
·
可压缩性:保留≥85%,优选地≥90%的初始厚度。为了测量可压缩性,使用以下方法。因此,当在材料上施加限定的压力时,测量厚度,并且随着时间的推移,测量厚度的减小。厚度测量用通用千分尺(frank pti gmbh)进行。使用面积为10cm2的圆形推杆作为样本架。将直径为47mm的圆形样品放置在板和推杆之间。对于可压缩性,测量厚度是通过将1kg重物放置在推杆顶部的同时监测样品的厚度5分钟来测量的。压缩率通过以下计算:
[0047][0048]
·
厚度和密度的均匀性在平均值附近至多10%变化。
[0049]
由于另外的增强材料的存在,与没有增强材料的实施方案相比,分离基质的x/y方向上的可加工性(卷料的冲洗、浸渍、干燥)可以有利地增加,因为在可能的生产/修改工艺(编织工艺)的拉伸方向上没有明显长度变化(伸长率)的力吸收可以增加。另外,增强材料(例如热塑性树脂)的材料的选择可以确保分离基质与色谱单元的热塑性装置部件的改善的连接。因此,可以通过另外的增强材料来改善整合色谱装置的可制造性。
[0050]
根据本发明的优选实施方案,第一连续相的中值桥直径比非圆柱形大直径连续连接的对流孔的中值孔径大0.4至4倍。该比值强烈地取决于对流孔隙率ε(例如,在ε=0.33的情况下(即对流孔隙率为33%),使得4.4
·
e-0.03
·
(ε
·
100)
相当于1.63的桥孔比),如以下等式所述。
[0051][0052]
如本文所用,中值桥直径被定义为来自科学出版物中的方法的所有孔数据的中位数,所述科学出版物为ley等人,journal of membrane science,第564卷,2018年,第543-551页,此时二元结构的相被反转(1变为0,反之亦然)(参见其第546-548页)。优选地,第一连续相的中值桥直径比非圆柱形大直径连续连接的对流孔的中值孔径大1至3倍,更优选地大1至2倍。在上述范围之外的比率可能负面地影响表面可及性,并且也因此负面地影响结合或处理时间。
[0053]
根据本发明的优选实施方案,当对流孔隙率ε是第二连续相的体积与第一和第二连续相的总体积之比时,对流孔隙率ε为0.05至0.7,更优选地为0.05至0.5,最优选地为0.1至0.4。对流孔隙率ε可以通过本领域技术人员已知的并且在guan,hong&guiochon,georges(1996)“study of physico-chemicalproperties of somepacking materials”(journal of chromatography a-j chromatogr a.731.27-40.10.1016/0021-9673(95)01197-8)中示例性显示的反相尺寸排阻色谱法测定。如本文所用,“第一和第二连续相的总体积”是指
第一相的体积和第二相的体积之和,而不考虑本发明的色谱材料中任选的惰性第三相(以及其体积)。如果对流孔隙率小于上述限定的范围,则色谱材料的渗透率可能由于色谱材料中存在较大量的多糖材料而降低。如果对流孔隙率大于上述限定的范围,则色谱材料的结合能力可能由于在色谱材料中存在较小量的多糖材料而降低。
[0054]
根据本发明的优选实施方案,本发明的色谱材料具有至少0.31
×
(ε
×
100)md(毫达西),更优选地至少1.52
×
(ε
×
100)md以及最优选地至少3.1
×
(ε
×
100)md的渗透率,其中ε是如上定义的对流孔隙率。因此,当对流孔隙率ε为0.33(即对流孔隙率为33%)时,本发明的色谱材料优选地具有至少10.2md,更优选地至少50.2md,并且最优选地至少102md的渗透率。渗透率强烈地取决于对流孔隙度ε。这可以通过几何考虑来得到。在恒定的中值孔径和更高的对流孔隙率下,必须存在更多数目的孔,因此在恒定压力下体积流量也较大。对于较小的对流孔隙,反之亦然。渗透率可以例如如科学出版物ley等人,journal of membrane science,第564卷,2018年,第543-551页(第547页:描述;第545页:测量)所述进行测定。渗透率越高,在使用色谱材料的色谱过程中所需的工艺压力越小。这有利地允许在色谱过程中更大的柱长和更大的床高,从而降低轴向色散。此外,渗透率的测定也是确认分离基质的双连续结构的方法,因为渗透率低于0.31
×
(ε
×
100)md(例如,在33%的对流孔隙率(即ε=0.33)下为10.2md)是不存在连续对流相的强烈指示。
[0055]
根据本发明的优选实施方案,非圆柱形大直径连续连接的对流孔的中值孔径的方差系数至多为0.8,更优选至多为0.7。如本文所用,中值孔径的方差系数是中值孔径的标准偏差除以中值孔径的平均值。中值孔径的“标准偏差”可以通过使用微软excel函数“stabw.n”来确定,它根据作为参数指定的总体来计算标准偏差(忽略逻辑值和文本)。标准偏差是值相对于其平均值(平均值)的离散度的度量。如果非圆柱形大对流孔的中值孔径的方差系数高于上述最大值,则色谱材料在色谱过程中的性能可能降低,即可能出现较早的突破曲线、较宽的洗脱峰和较小的色谱分辨率。
[0056]
根据本发明的优选实施方案,第一连续相的中值桥直径的方差系数至多为0.7,更优选至多为0.6。如本文所用,中值桥直径的方差系数是中值桥直径的标准偏差除以中值桥直径的平均值。中值桥直径的标准偏差的测定类似于上述中值孔径的标准偏差的方法。如果第一连续相的中值桥直径的方差系数高于上述最大值,则可能发生分离基质的结合位点不能被充分利用,导致色谱过程中更早的突破和更宽的洗脱峰。
[0057]
根据本发明的进一步优选的实施方案,本发明的色谱材料的分离基质在第一和第二连续相之间具有至少0.04m2/ml(其中体积ml是指没有任选的第三惰性相/增强材料的分离基质体积),更优选大于0.1m2/ml,并且最优选大于0.2m2/ml的特定界面。如果弯曲度是2.5,则第一和第二连续相之间的特定界面可以通过以下等式来测定。如果弯曲度不同于这种示例性但典型的弯曲度,则渗透率会通过一个因素变化,然后观察到的弯曲度从2.5到较高的渗透率(对于较低的弯曲度)或较低的渗透率(对于较高的弯曲度),如本领域技术人员已知的并且在以下等式中给出的(同样参见科学出版物berg等人,transport in porous media,2014,第10页,https://arxiv.org/pdf/1505.02424.pdf)。
[0058]
界面
特定
=(-37.868∈5+75.428∈
4-66.017∈3+34.08∈
2-2.7346∈+0.1333)
·
渗透率-0.5
[0059][0060]
如果第一和第二连续相之间的特定界面低于0.04m2/ml,则转变成小直径扩散孔的界面可能太小,使得较少的组分(目标物质和/或污染物)可以扩散到第一连续相中。因此,在扩散时间保持不变的同时,可以限制传质和因此的结合能力,从而导致结合能力变小或处理时间变长。
[0061]
色谱材料的形状,与任选的第三惰性相的存在无关,没有特别限制。因此,色谱材料可以是例如平板(膜)或中空纤维膜、棒状(monolith)或颗粒的形状。
[0062]
本发明的另一方面涉及生产本发明的色谱材料的方法。上述每个实施方案也适用于通过本发明的方法生产的色谱材料。
[0063]
特别地,提供了生产本发明的包括基于聚合物网络材料的自支撑双连续分离基质的色谱材料的方法,其中所述第一连续相由自胶凝多糖形成,所述方法包括以下步骤:
[0064]
(a)制备包括自胶凝多糖和第一溶剂的溶液(a);
[0065]
(b)制备包括至少一种表面活性剂和第二溶剂的溶液(b),所述第二溶剂是与溶液(a)不混溶的溶剂;
[0066]
(c)将溶液(a)和溶液(b)合并以产生合并的溶液(c);
[0067]
(d)在使自胶凝多糖保留在作为溶液(a)的第一溶剂中的溶液的条件下乳化合并的溶液(c)以获得乳液,所述乳液呈溶液(a)包溶液(b)的乳液的形式;以及
[0068]
(e)使含有自胶凝多糖的溶液(a)在一定条件下通过凝胶化固化以形成色谱材料,所述色谱材料包括分离基质,所述分离基质包括自胶凝多糖作为第一连续相。
[0069]
在本发明方法的步骤(a)中,制备包括自胶凝多糖和第一溶剂的溶液(a)。第一溶剂没有特别限制,并且可以使用自胶凝多糖可溶于其中(在室温下或在高温下)的任何溶剂。第一溶剂也可以是两种或更多种这样的溶剂的混合物。根据优选的实施方案,第一溶剂是水。溶液(a)可以通过将自胶凝多糖和第一溶剂搅拌和加热(至例如30℃或更高、40℃或更高、50℃或更高、60℃或更高、70℃或更高、80℃或更高、或90℃或更高)预定时间(例如2分钟或更多、5分钟或更多、10分钟或更多、15分钟或更多)直到自胶凝多糖(完全)溶解在第一溶剂中来制备。
[0070]
自胶凝多糖的浓度没有特别限制。根据优选实施方案,溶液(a)中多糖的浓度为0.5至8wt%,优选地为1至6wt%,并且最优选地为2.5至5wt%。如果浓度低于上述范围,则自胶凝多糖的胶凝能力可能不够,这可能导致多糖凝胶的稳定性变差。如果浓度高于上述范围,则可能发生在色谱过程中待结合的目标组分的不希望的尺寸排阻和/或极低的扩散传质到凝胶中。
[0071]
在本发明方法的步骤(b)中,制备包括至少一种表面活性剂和第二溶剂的溶液(b),所述第二溶剂是与溶液(a)不混溶的溶剂。如本文所用,“不混溶的”意指在70℃下溶剂在另一种溶剂中的溶解度小于100g/l,优选地在70℃下小于50g/l,并且最优选地在70℃下小于10g/l。
[0072]
第二溶剂可以是单一溶剂或两种或更多种溶剂的混合物。第二溶剂中的至少一种
溶剂是与溶液(a)不混溶的溶剂。根据本发明的优选实施方案,第二溶剂是选自c
4-12
醇(例如1-己醇、2-庚醇、1-辛醇、1-壬醇、1-癸醇、1-十二烷醇)、c
4-12
异醇、烷烃(例如辛烷和癸烷)、粘度为4-200cp的硅油,以及粘度为4-200cp的天然油或其混合物中的一种或多种。硅油可以例如是聚二甲基硅氧烷(例如来自wacker的硅油20cp)。天然油可以例如是橄榄油、葵花油或菜籽油。第二溶剂的特别优选的实施方案是辛烷、癸烷、1-辛醇、1-癸醇和1-十二烷醇。最优选的第二溶剂是癸醇,特别是1-癸醇。溶液(b)可以例如通过在室温或在高温(例如30℃或更高、35℃或更高、40℃或更高、或50℃或更高)下搅拌预定时间(例如2分钟或更多、5分钟或更多、10分钟或更多、15分钟或更多)来制备。
[0073]
根据本发明的优选实施方案,至少一种表面活性剂具有8至16,优选地10至14,并且最优选地11至13的hlb值。表面活性剂的hlb(亲水-亲油平衡)是其亲水或亲油程度的量度,通过计算分子的不同区域的值来测定,如griffin("classification of surface-active agents by'hlb'",journal of the society of cosmetic chemists,1949,1(5):311
–
26);以及“calculation of hlb values of non-ionic surfactants”,journal of the society of cosmetic chemists,1954,5(4):249
–
56)和davies(“a quantitative kinetic theory of emulsion type,i.physical chemistry of the emulsifying agent",gas/liquid and liquid/liquid interface,proceedings of the international congress of surface activity,1957,pp.426
–
38)所述。取单一表面活性剂的hlb值的列表或计算值,混合物的hlb值由例如在以下针对两种表面活性剂的混合物的公式中的质量分数确定,其中m是所使用的表面活性剂的质量,以及hlb是特定表面活性剂的列表或计算的hlb值。
[0074][0075]
hlb值越小,所得对流孔径和所得桥直径越小。hlb值是关于多糖相(第一连续相)的体积和多糖浓度的独立参数。如果两个连续相(第一连续相和第二连续相)的体积比用于控制孔径,则它还改变对流孔隙率并因此改变渗透率。如果多糖浓度用于控制第二连续相(孔相)的孔径,则第一连续相(多糖相/扩散相)也被改变。例如,当使用1-癸醇作为有机溶剂时,在较高的多糖浓度下,对流孔变得更大,并且同时扩散孔变得更小,这可能导致排除较大的分子和降低的扩散传质。相反地,再次使用1-癸醇作为有机溶剂,在较低的多糖浓度下,对流孔变得更小并且扩散孔变得更大,这不仅改善了传质,而且还减小了用于在色谱过程中目标组分与分离基质可能结合的表面积。通过使用上述hlb值范围,可以有利地影响对流孔径而没有这些效果。
[0076]
鉴于上述情况,至少一种表面活性剂的hlb值优选在上述hlb值范围内,并且至少一种表面活性剂还能够产生双连续结构。此外,至少一种表面活性剂的使用优选地在乳液中的两个液相之间产生多达1mn/m的界面张力,并且优选地是非离子的。用测角器oca 15pro和悬滴法测量两相之间的表面张力。将填充有液相a(示例性水,但所有其它溶液和混合物都是可能的)的注射器的针置于液相b(示例性与水不混溶的任何有机溶剂,但所有其它溶液和液体混合物都是可能的)中。相a的液滴以例如10μl的体积或以例如1μl/s的分配速率连续地分配在相b中,并且用相对于针90
°
放置的照相机记录。根据液滴的曲率和针的直径,用来自dataphysics instruments的软件sca 22-表面/界面张力计算相a和相b之间
的表面张力(参见f.k.hansen,g.rodsrud,surface tension by pendant drop,journal of colloid and interface science,第141卷,第1期,1991)。
[0077]
优选的表面活性剂是山梨醇的聚山梨醇酯和/或c
2-150
脂肪酸酯,诸如例如商业上可获得的吐温20/60/80(吐温20:聚氧乙烯脱水山梨醇单月桂酸酯;吐温80:聚氧乙烯脱水山梨醇单油酸酯)或span 20/60/80/85。根据本发明,可以使用一种单一表面活性剂或者两种或更多种表面活性剂的组合。根据特别优选的实施方案,组合使用两种不同的表面活性剂。
[0078]
为了制造双连续结构(而不是泡沫结构),根据所用的有机溶剂和多糖浓度,必须使用最小的表面活性剂浓度。例如,基于溶剂(即第一和第二溶剂两者)和表面活性剂的总体积的体积比,表面活性剂的优选量由下面给出的公式计算并且仅描述了至少存在于溶液(b)中的表面活性剂的百分比,并且优选量对于1-十二烷醇为至少2.5vol%、对于1-癸醇为至少4.5vol%、对于1-壬醇为至少7.5vol%、对于1-辛醇为至少7.5vol%、对于1-己醇为至少15vol%、对于2-庚醇为至少7.5vol%、对于环己烷为至少2vol%、对于辛烷为至少3vol%、对于癸烷为至少2.5vol%以及对于硅油(pdms)为至少7vol%。因此,该表面活性剂浓度是根据下列方程所用的有机相的总量来计算的。有机相的总量越高,所需的表面活性剂的总量越高。
[0079][0080]
上述表面活性剂浓度(取决于有机溶剂)特别优选地在溶液(a)中多糖的浓度为2.5至5wt%的情况下使用。
[0081]
在本发明方法的步骤(c)中,将溶液(a)和溶液(b)合并以产生合并的溶液(c)。步骤(c)可以在室温或高温下进行,例如30℃或更高、35℃或更高、40℃或更高、或50℃或更高。根据优选实施方案,步骤(c)中的合并溶液(c)中的第二溶剂的体积比为10至70vol%,因为在该范围内,可以在本发明的步骤(d)中产生合适的乳液。
[0082]
在本发明方法的步骤(d)中,在使自胶凝多糖保留在作为溶液(a)的第一溶剂中的溶液的条件下乳化合并的溶液(c)以获得乳液,所述乳液呈溶液(a)包溶液(b)的乳液的形式。根据本发明的优选实施方案,步骤(d)通过将合并的溶液(c)暴露于通过例如使用turrax specification:t25digital ultra-model t25d,firma ika(在10.000至20.000rpm下)或来自ika的具有类型r1303 dissolver stirrer分散盘的eurostar 40digital(在1.000rpm至2.000rpm下)(剧烈)搅拌、分散或均化而产生的剪切力来进行。剧烈搅拌、分散或均化有利地使得能够产生中值孔径为0.1μm至6μm的非圆柱形大直径连续连接的对流孔。
[0083]
在本发明方法的步骤(e)中,使含有自胶凝多糖的溶液(a)在一定条件下通过凝胶化而固化以形成色谱材料,所述色谱材料包括分离基质,所述分离基质包括自胶凝多糖作为第一连续相。使自胶凝多糖在一定条件下通过凝胶化而固化是例如通过将溶液(a)包溶液(b)的乳液冷却至低于多糖的胶凝温度的温度来实现的,例如冷却至至多45℃、至多40℃、至多35℃、至多30℃或至多25℃。在其他实施方案中,固化例如通过向溶液(a)包溶液(b)的乳液中加入例如钾盐来实现。根据本发明的优选实施方案,步骤(e)通过将乳液倾倒
在铸型(casting form)上并在胶凝点以下冷却乳液来进行。如本文所用,“铸型”是可以在其上提供/倾倒乳液以固化的任何表面,并且优选地该表面是可以快速冷却的金属表面,并且最优选地该表面在0℃至100℃的范围内是可加热的和可冷却的。冷却速率没有特别限制,并且通常使用0.01℃/秒至50℃/秒的冷却速率。根据本发明的优选实施方案,冷却速率为0.2℃/秒至30℃/秒,特别优选为1℃/秒至20℃/秒。
[0084]
在色谱材料还打算包括用作增强材料的第三惰性相的情况下,第三惰性相优选存在于本发明方法的步骤(e)中以将第三惰性相(随机)引入分离基质中。也就是说,优选将步骤(d)的溶液(a)包溶液(b)的乳液倾倒在步骤(e)中的增强材料(第三惰性相)上,并使溶液(a)在第三惰性相存在下通过凝胶化而固化。
[0085]
根据本发明方法的特别优选的实施方案,自胶凝多糖是琼脂糖,并且该方法包括以下步骤:
[0086]
(a)通过将水包琼脂糖悬浮液加热到至少90℃持续至少15分钟以将琼脂糖溶解在水中来制备包括琼脂糖和水的溶液(a);
[0087]
(b)制备包含所述至少一种表面活性剂和所述第二溶剂的溶液(b),所述第二溶剂是水不混溶性有机溶剂,并且将溶液(b)加热至高于所用琼脂糖的凝胶化温度的温度(例如至少40℃);
[0088]
(c)在高于所用琼脂糖的凝胶化温度的温度(例如至少40℃)下合并溶液(a)和溶液(b)以产生合并的溶液(c);
[0089]
(d)在高于所用琼脂糖的凝胶化温度的温度(例如至少40℃)下乳化合并的溶液(c),以使琼脂糖保留在作为溶液(a)的第一溶剂中的溶液中,以获得乳液,所述乳液呈溶液(a)包溶液(b)的乳液的形式;以及
[0090]
(e)使含有自胶凝琼脂糖的溶液(a)在低于所用琼脂糖的凝胶化温度的温度(例如,低于35℃)下通过凝胶化而固化,以形成包括分离基质的色谱材料,所述分离基质包括自胶凝琼脂糖作为第一连续相。
[0091]
可任选地通过引入官能团和/或配体,诸如例如阴离子配体、阳离子配体、疏水配体、混合模式配体(例如两种或更多种官能团,例如阳离子官能团和疏水官能团)、亲和配体等来修饰分离基质。通过引入官能团和/或配体的这种修饰为本领域技术人员所熟知。
[0092]
根据本发明,有利的是可以提供色谱材料,其包括具有扩散孔和对流孔的基于聚合物网络材料的自支撑双连续分离基质,当与现有技术中已知的分离基质相比时,对流孔具有非常小的尺寸。由于小的对流孔,扩散路径长度也相当小,并且在非常短的扩散时间内可进入分离基质的多孔桥内的扩散孔。此外,与纯对流基质相比,本发明的色谱材料由于其特定结构而显示出高渗透率,因此当在色谱过程中使用色谱材料时导致较小的工艺压力。这有利地使色谱过程中较长的柱长和较低的轴向分散。此外,与现有技术的材料相比,本发明的色谱材料有利地显示出较低的结垢倾向。鉴于上述情况,本发明的色谱材料通过优异的的结合位点可及性、后期突破曲线和窄洗脱峰,有利地使操作色谱过程非常快速并且有效,并且因此本发明的分离基质特别适用于需要高结合能力的扩散限制过程,如例如在大蛋白、病毒颗粒等的结合和洗脱应用中观察到的。
[0093]
附图显示了:
[0094]
图1示出了多糖颗粒和多糖膜中的对流和扩散流动路径的比较。
[0095]
图2示出了用于双连续和闭孔泡沫结构(实施例1)的对流孔的渗透率和中值直径(在ε=0.33对流孔隙率下)之间的关系。
[0096]
图3示出了在根据us5,723,601a(实施例1)中所述的反应条件(工艺参数)制备色谱材料中,中值孔径和所用表面活性剂浓度(在ε=0.33对流孔隙率下)的关系。
[0097]
图4示出了对于表2的选定实施例(实施例2)的非圆柱形大直径连续连接的对流孔的中值桥直径和中值孔径(在ε=0.33对流孔隙率下)的比例。
[0098]
图5示出了用于测定dbc 10%的装置。
[0099]
图6示出了分离基质的中值桥直径与动态结合能力(在ε=0.33对流孔隙率下)之间的关系(实施例3)。
[0100]
图7示出了第一连续相的中值桥直径的方差系数与动态结合能力(在ε=0.33对流孔隙率下)之间的关系(实施例4)。
[0101]
图8示出了第一和第二连续相之间的特定界面与动态结合能力(在ε=0.33对流孔隙率下)之间的关系(实施例5)。
[0102]
图9示出了取决于停留时间的dbc 10%(实施例8)。
[0103]
图10示出了本发明色谱材料和现有技术材料的dbc 10%作为停留时间的函数的比较(实施例8)。
[0104]
图11示出了在用不同的清洁策略(负荷:mab浓度为2.8mg/ml的澄清细胞培养物中的mab)循环期间的反式装置压力(实施例9)。
[0105]
图12示出了三个不同色谱模块的单个结合和洗脱循环的叠加色谱图(实施例10)。
[0106]
图13示出了在280nm处具有mab a,uv信号的200个循环的覆盖图(实施例11)。
[0107]
图14示出了在280nm处具有mab b,uv信号的200个循环的覆盖图(实施例11)。
[0108]
图15示出了在280nm处具有mab c,uv信号的200个循环的覆盖图(实施例11)。
[0109]
图16示出了用mab a,δ压力[mpa]进行200个循环的覆盖图(实施例11)。
[0110]
图17示出了用mab b,δ压力[mpa]进行200个循环的覆盖图(实施例11)。
[0111]
图18示出了用mab c,δ压力[mpa]进行200个循环的覆盖图(实施例11)。
[0112]
图19示出了针对mab a、b和c的产率vs.循环数(实施例11)。
[0113]
图20示出了针对mab a、b和c的hcp去除vs.循环数(实施例11)。
[0114]
图21示出了针对mab a、b和c的dna去除vs.循环数(实施例11)。
[0115]
图22示出了针对mab a、b和c的proa配体浸出vs.循环数(实施例11)。
[0116]
图23示出了针对mab a、b和c的洗脱峰宽vs.循环数(实施例11)。
[0117]
图24示出了针对mab a、b和c的池浓度vs.循环数(实施例11)。
[0118]
图25示出了针对mab a、b和c的聚集浓度vs.循环数(实施例11)。
[0119]
图26示出了本发明的3层和12层色谱装置的突破行为(实施例12)。
[0120]
图27示出了本发明的3层和12层色谱装置的洗脱峰形状(实施例12)。
[0121]
图28示出了作为ciex材料的停留时间的函数的dbc 10%结果(实施例13)。
[0122]
图29示出了作为mm材料的停留时间的函数的dbc 10%结果(实施例13)。
[0123]
本发明将在以下实施例中进一步说明,但不限于此。
实施例
[0124]
一般制备例:
[0125]
制备自胶凝多糖溶液(例如3%琼脂糖水溶液),并在高温(例如95℃)下储存直至完全溶解。将癸醇(例如27.39g)、吐温80(例如2.48g)和span80(例如0.98g)混合并在高温(例如60℃)下搅拌以形成有机相。将上述自胶凝多糖溶液(例如,量为63.72g的琼脂糖溶液)在50℃下倾倒入在水桶中回火的烧杯中,并且在搅拌的同时缓慢加入有机相。用ultraturrax(优选地)在20,000rpm和70-75℃下剧烈搅拌乳液10分钟。可选地,用来自ika的eurostar 40 digital和类型r1303 dissolver stirrer的分散盘(优选地以2.000rpm)剧烈搅拌乳液。在乳化过程期间观察温度,并且如果需要,将水桶冷却或加温以保持乳液温度恒定。同时,准备好了浇铸平台。为此,任选地,将用作增强材料的羊毛片(a sheet of fleece)(第三惰性相)粘接到浇铸平台的两端,其通过来自下方的温水流在40℃下进行回火。在没有第三惰性相(增强材料/羊毛)的情况,同样的过程也是有效的。将乳液倾倒在浇铸平台的一端上(在羊毛存在下,在羊毛顶部)并且用刮刀铺展以获得具有例如300μm的宽度的乳液层。立即使用来自下方的冷自来水将浇铸平台冷却至15℃。由此,诱导了琼脂糖的凝胶化。在等待1至2分钟后,乳液变成坚固的膜(任选附着到羊毛上)。将具有或没有增强材料/羊毛的膜从浇铸平台上小心地取下,并放置于具有自来水的桶中以洗出有机相。在用自来水洗涤膜20分钟后,将其在异丙醇(2
×
10分钟)和水/异丙醇混合物(50:50,2
×
10分钟)中轻轻摇动,每个步骤包括改变洗涤介质。最后,将膜在自来水中再洗涤20分钟。
[0126]
对于共焦显微镜分析,没有增强材料/羊毛的样品被证明是获得良好显微镜图像的最容易的方法。为此,在浇铸乳液之前,在羊毛中切出直径为13mm的小孔。浇铸过程与上述相同。在洗涤过程之前,在同一孔处切下膜,获得没有羊毛的膜。
[0127]
亲和配体(蛋白质a)的配体固定在两步法中进行。在第一步骤中,在基质具有与环氧乙烷反应的官能团的情况下,用双环氧乙烷分子进行活化,使得双环氧乙烷的至少一个环氧乙烷基团与基质反应。然后在基质表面上如此产生的未反应的环氧乙烷可用于进一步的表面化学。
[0128]
第二步骤是通过将配体加入到活化的基质中来进行的,所述活化的基质具有至少一个相对于环氧乙烷的反应性侧。这种或类似的可能方法可以在a.krishna mallia,paul k.smith,greg t.hermanson,immobilized affinity ligand techniques,elsevier science,1992中找到。
[0129]
实施例1:根据本发明和现有技术的色谱材料的制备和表征根据上述一般制备例,在表1所示的条件下制备色谱材料样品编号1至33。通过将所用蛋白质a配体溶解在浓度为10mg/ml,ph为7的1m kpi缓冲液中,引入亲和配体。在制备该偶联溶液后,在室温下将分离基质插入该偶联溶液中至少16小时。此后,将分离基质用0.1m 1
×
pbs缓冲液,ph7洗涤并储存在其中直到使用。
[0130]
在该实施例和以下实施例中,在以下方面表征色谱材料:
[0131]
·
配体密度:
[0132]
蛋白质a材料:bca测定,文献:c.m.stoscheck:quantitation of protein.in:methods in enzymology.band 182,1990,s.50
–
68,pmid 2314256
[0133]
iex材料:用本领域技术人员已知的例如naoh或hcl滴定。
[0134]
·
sbc(静态结合能力)的测定:
[0135]
sbc值通过将目标分子在结合缓冲液条件下孵育12h和在洗脱条件缓冲液中洗涤目标分子至少1h后的洗脱步骤来测量。在sbc测量之前测量的校准曲线内,通过在280nm处的uv-vis测量洗脱缓冲液中的浓度。
[0136]
·
dbc 10%的测定(在10%突破时的动态结合能力):用来自ge-的avant150色谱系统测量dbc 10%。首先,将所述材料放入内部测量装置(lp15,图5)中,并通过鲁尔锁(luer-lock)连接到avant150。然后将至少20个分离基质体积(mv)的结合缓冲液通过分离基质进行净化。然后将具有溶解的目标蛋白(1mg/ml)的结合缓冲液通过分离基质净化,直至达到初始目标蛋白溶液的浓度的10%(0.1mg/ml)。在此之前以ml计的净化体积是由于1mg/ml的浓度等同于以mg计的质量,并且通过除以分离基质体积得到dbc 10%,从而得到以mg/ml计的dbc 10%。
[0137]
·
dbc 100%的测定(在100%突破时的动态结合能力):用来自ge-的avant150色谱系统测量dbc 100%。首先,将所述材料放入内部测量装置(lp15,图5)中,并通过鲁尔锁连接到avant150。然后将至少20个分离基质体积(mv)的结合缓冲液通过分离基质进行净化。然后将具有溶解的目标蛋白(1mg/ml)的结合缓冲液通过分离基质净化,直至达到初始目标蛋白溶液的浓度的100%(1mg/ml)。然后可以对这样得到的突破曲线下的面积进行积分,然后可以通过测量的面积、达到初始浓度的100%所需的体积和以mg/ml计的分离基质体积来确定dbc 100%。
[0138]
表2中显示了分离基质的表征结果。
[0139]
[0140]
[0141]
[0142][0143]
样品编号1至15是根据us 5,723,601 a中描述的反应条件(工艺参数)制备的色谱材料。样品编号1至10是双连续分离基质,但是每个分离基质的非圆柱形大直径连续连接的
对流孔具有大于6μm的中值孔径。从样品编号1至5可以看出,较少的表面活性剂(在恒定的多糖浓度下)导致对流孔的较小孔径。然而,在表面活性剂的浓度降至某一阈值以下的样品编号11至15中,获得泡沫结构而不是双连续结构。泡沫由部分或多或少连接的球状结构组成,作为扩散相之间的对流相(大孔),即大孔不互连。固有地较差的连接性导致结构在相当的孔径(即对流孔的中值直径)下具有非常低的渗透率,因为没有经由这些孔的通流。图2示出了双连续和泡沫结构的对流孔的渗透率和中值直径之间的关系。因此,泡沫结构只能以非常不均匀和不适当的方式穿过,这导致流体在色谱介质中均匀分布的缺点,以及除了较低的渗透率之外,由于不均匀的流动以及因此不均匀的结合位置可及性而导致的动态结合的缺点。
[0144]
图3示出了在根据us 5,723,601a中所述的反应条件(工艺参数)制备色谱材料中,中值孔径和所用表面活性剂浓度的关系。在us 5,723,601a中所述的反应条件下,不可能制备具有非圆柱形大直径连续连接的对流孔的双连续分离基质,所述对流孔具有0.1μm至6μm的中值孔径。
[0145]
样品编号16至27是根据本发明的色谱材料,其包括具有非圆柱形大直径连续连接的对流孔的双连续分离基质,所述对流孔具有0.1μm至6μm的中值孔径。特别是当与具有相似对流孔的中值孔径的样品编号11至15的泡沫相比时,分离基质显示出优异的渗透率。本发明的色谱材料在10%突破时具有优异的结合位点可及性和动态结合能力。
[0146]
样品编号28至33是如比较例所制备的色谱材料,其中使用与(本发明)样品编号16至20和27中相同的溶剂,但是在制备中所用表面活性剂的浓度已经降低到低于某一阈值,从而形成泡沫结构。样品编号28至33具有类似于样品编号16至27的中值孔径的对流孔,但样品编号28至33的泡沫结构的对流孔不像样品编号16至27的对流孔那样连续连接(互连)。因此,当将样品编号28至33的泡沫结构与样品编号16至27的双连续结构进行比较时,泡沫结构在相当的对流孔径下显示出降低的渗透率。此外,当与样品编号18至27的双连续结构相比时,样品编号30至33的泡沫结构在10%突破时显示出较差的动态结合能力。
[0147]
实施例2:大直径对流孔的平均桥直径与平均孔径之间的关系
[0148]
制备包括自胶凝多孔多糖作为第一连续相的分离基质并分析大直径对流孔的平均桥直径和平均孔径。结果描绘在图4中,其清楚地显示了大直径对流孔的平均桥直径和平均孔径的比例。特别地,在恒定的孔体积分数(对流孔隙率)的情况下,大直径对流孔变得越大,桥变得越大,因为桥仅代表大直径对流孔之间的空间。
[0149]
实施例3:分离基质的dbc 10%与它们的中值桥直径之间的关系
[0150]
制备包括自胶凝多孔多糖作为第一连续相的分离基质,并分析它们的中值桥直径和dbc 10%(动态结合能力10%突破(配体:亲和蛋白质a;缓冲液:磷酸盐缓冲盐水(pbs)ph7.3;在pbs中的分析物1mg/ml单克隆抗体(mab);速度:5mv/min=12秒在介质上停留或与介质接触的时间)。所有数据通过使用如图5所示的装置(称为lp15)内平板材料得到,并且用avant150测量。
[0151]
结果描绘在图6中,其清楚地显示了当保持工艺参数恒定(接触和工艺时间)时,分离基质的中值桥直径越大,动态结合能力也越小。
[0152]
实施例4:分离基质的dbc 10%与它们的第一连续相的中值桥直径的方差系数之间的关系
[0153]
制备包括自胶凝多孔多糖作为第一连续相的分离基质,并分析它们的第一连续相的中值桥直径的方差和dbc 10%(配体:亲和蛋白质a;缓冲液:磷酸盐缓冲盐水(pbs)ph7.3;在pbs中的分析物1mg/ml单克隆抗体(mab);速度:5mv/min=12秒在介质上停留或与介质接触的时间)。
[0154]
结果描绘在图7中,其清楚地显示了当保持工艺参数恒定(接触和工艺时间)时,中值桥直径的方差系数越大,动态结合能力也越小。
[0155]
实施例5:分离基质的dbc 10%与它们在第一和第二连续相之间的特定界面之间的关系
[0156]
制备包括自胶凝多孔多糖作为第一连续相的分离基质,并分析它们在第一和第二连续相之间的特定界面和dbc 10%(配体:亲和蛋白质a;缓冲液:磷酸盐缓冲盐水(pbs)ph7.3;在pbs中的分析物1mg/ml单克隆抗体(mab);速度:5mv/min=12秒在介质上停留或与介质接触的时间)。本实施例中的分离基质被设计成具有经由配体固定可达到的最大可能静态结合能力。
[0157]
结果描绘在图8中,其清楚地显示了当保持工艺参数恒定(接触和工艺时间)时,第一和第二连续相之间的特定界面越大,传质可以发生得越快,并且动态结合能力也越大。
[0158]
实施例6:
[0159]
包括作为第一连续相的自胶凝多孔琼脂糖的本发明色谱膜(样品编号34和35)根据上述一般制备例在表3中所示的以下条件下制备。
[0160]
特别地,制备自胶凝琼脂糖溶液,并在高温(例如95℃)下储存直至完全溶解。将1-癸醇、吐温80和span80混合并在高温(例如80℃)下搅拌以形成有机相。将上述自胶凝琼脂糖溶液倾倒入在高温(例如80℃)下回火的容器中,并且在搅拌的同时缓慢加入有机相。用disperser ystral inline z66(c)以例如5000rpm剧烈分散乳液。将用作增强材料的第三惰性相(例如羊毛)附着到浇铸平台,所述浇铸平台保持低于胶凝温度的温度(例如30℃)。将乳液作为薄膜浇铸到浇铸平台上,浸泡羊毛。由于浇铸平台的温度低于琼脂糖的胶凝温度,琼脂糖立即开始形成胶凝的本发明的色谱膜。将本发明的具有增强材料的色谱膜从浇铸平台上取下,卷绕在辊上,并用异丙醇冲洗,最后用水冲洗。
[0161][0162]
实施例7:在典型的操作压力下第三惰性相(增强材料)的压缩对本发明的色谱膜的渗透率的影响
[0163]
根据实施例6,使用不同的平板增强材料作为制备本发明的色谱膜(样品编号34和35)的第三惰性相。使用的增强材料是通过压缩测量的具有不同压缩率的羊毛材料a、b和c,如通过上述等式计算的。对于压缩测量,是通过将1kg的重物放置在推杆顶部测量在压缩之前和之后的厚度,同时在20秒之后监测样品的厚度。
[0164]
如科学出版物ley等人,journal of membrane science,第564卷,2018,第543-551页(第547页:描述;第545页:测量)所述测定所得本发明色谱膜的渗透率χ。将单层本发明色谱膜在100毫巴跨膜压力(tmp)下的渗透率与在100毫巴tmp下本发明色谱膜的3层的叠层的渗透率进行比较。通过以下计算将渗透率归一化为不同的厚度:
[0165][0166]
这里,ν是水通过膜单层或叠层的流速(以计),μ是水的粘度(以mpa
·
s计)以及δx是单层或叠层的厚度(以μm计)。
[0167]
本发明的色谱膜的相对渗透率通过如下计算
[0168][0169]
表4
[0170][0171]
第三惰性相的压缩在典型的操作tmp下对本发明的色谱膜的渗透率具有强烈的影响。第三惰性相的压缩越高,本发明色谱膜的3层叠层在典型操作压力下的相对渗透率越高。
[0172]
实施例8:用亲和蛋白质a配体官能化的本发明色谱膜的性能特征。
[0173]
根据实施例6所述,产生增强的本发明的色谱膜(=样品a)。具有根据实施例7所述的第三惰性相c的样品编号34根据实施例1所述进行改性。所产生的本发明的色谱膜样品a表现出以下材料和性能特征:厚度为280μm,渗透率为66md,配体密度为19.8g/l。将本发明的色谱膜样品a切成圆形试样并且组装成可重复使用的过滤装置中的4层圆形膜叠层,所述过滤装置具有聚丙烯过滤器壳体(过滤器台和过滤器盖),所述过滤器壳体具有鲁尔锁连接器并且由不锈钢保持器机械地支撑,所述不锈钢保持器在过滤器盖、膜叠层和过滤台外形成机械稳定的色谱单元。膜叠层的正面面积为5cm2,厚度为1.12mm,这导致总膜体积(mv)为0.56ml。
[0174]
使用avant 150色谱系统(cytiva,28976337)测定本发明的色谱膜装置在不同停留时间对纯化的单克隆抗体的动态结合能力。用于性能表征的平衡和洗涤缓冲液由磷酸盐缓冲盐水(0.1m ph7.4,电导率16ms/cm)制成;使用ph2.9的0.1m浓度的乙酸作为洗脱缓冲液。通过cho细胞发酵产生单克隆抗体(mw~140kda)。通过深度过滤(sartoclear dl20 0.8m
2 29xdl20-fcc;sartoclear dl90 0.8m
2 29xdl90-fcc)完成发酵液的澄清,并通过0.2μm无菌过滤器胶囊(sartopore 2xlg 30“5447307g3—ss)进行无菌过滤。使用protein a mabselectsure柱(mabselectsure ge 17-5438;xk 50/20ge)纯化抗体。用于膜的性能表征的负载溶液由含有1mg/ml纯化抗体的平衡缓冲液制备。用于动态结合能力测定的流速为1、3、5和10mv/min,这对应于0.1、0.2、0.3和1分钟的停留时间。通过在线uv检测器在280nm测定流通级分中抗体的浓度,抗体的已知消光系数为1.42。基于uv信号计算负载溶液的10%突破的动态结合能力(=dbc 10%;参见图9)。
[0175]
使用0.4ml单位的fibro hitrap prisma(cytiva 17549856 lot 17103843)作为代表现有技术的纯对流蛋白质a亲和色谱材料的基准色谱材料,其不显示扩散相。所述材料由纤维素纳米纤维制成,其中配体固定在纤维的表面上,因此通过纤维材料的对流可直接进入,消除了任何相关的扩散传输限制。
[0176]
使用1ml mabselectsure柱(cytiva,11003493)作为代表现有技术蛋白质a亲和树脂材料的另外的基准材料。所述树脂由交联的琼脂糖制成并且具有85μm的平均珠粒尺寸(ge instructions 71-5020-91 ac),代表扩散相。
[0177]
对两种现有技术的蛋白质a亲和材料和本发明的蛋白质a色谱材料的色谱性能进行了表征。如前所述测定数据。用于性能表征的平衡和洗涤缓冲液由磷酸盐缓冲盐水(0.1m ph 7.4,电导率16ms/cm)制成。使用ph2.9的0.1m浓度的乙酸作为洗脱缓冲液。通过cho细胞发酵产生单克隆抗体(mab)(mw 140kda)。发酵液的澄清是通过深度过滤(sartoclear dl20 0.8m
2 29xdl20-fcc;sartoclear dl90 0.8m
2 29xdl90-fcc),然后通过0.2μm无菌过滤器胶囊(sartopore 2xlg 30“5447307g3—ss)进行无菌过滤实现的。通过使用proa mabselectsure柱(mabselectsure ge 17-5438;xk 50/20ge)纯化抗体。
[0178]
用于本发明色谱膜的性能表征的负载溶液由含有1mg/ml纯化mab的平衡缓冲液制备。通过在线uv检测器在280nm测定流通级分中mab的浓度,抗体的已知消光系数为1.42。基于uv信号(=dbc 10%)计算负载溶液的10%突破的动态结合能力。
[0179]
如图10所示,与现有技术的琼脂糖树脂材料相比,本发明的色谱膜显示出明显更高的dbc 10%vs.停留时间性能,使得能够进行高度生产和快速的抗体捕获过程。动态结合性能与现有技术的对流膜材料非常相似。对于非常短的停留时间(《0.2分钟),对流材料显示出稍微较高的dbc 10%值。
[0180]
实施例9:与具有类似结合能力的纯对流材料相比,本发明的色谱膜的低结垢倾向
[0181]
根据实施例6,产生增强的本发明的色谱膜,具有根据实施例7所述的第三惰性相c的样品编号34根据实施例1所述进行改性。将如实施例8中所述组装成4层色谱装置配置的新制备的膜的结垢倾向与如实施例8中所述的未使用的纯对流基准材料fibro(cytiva 17549856lot 17103843)的结垢倾向进行比较,以比较这些不同色谱材料的结垢倾向。如实施例8所述,使用由cho细胞产生的澄清mab进料流进行多次结合和洗脱循环,观察通过在连续结合和洗脱循环期间观察反式装置压力的变化所指示的材料的渗透率变化。
[0182]
进料流中的抗体浓度为2.8mg/ml。在进行循环实验之前,用如实施例8中产生的纯化mab测定所用装置的动态结合能力。值得注意的是两种材料在0.2分钟的停留时间内具有约40mg/ml的相似结合能力(参见图10)。然后将循环研究的负载设定为测定的dbc 10%值的80%。结合和洗脱循环中的不同阶段如下:1)用8mv平衡缓冲液以10mv/min平衡;2)用澄清进料流以5mv/min装载至dbc 10%的80%(即7.5ml的cytiva hitrap 0.4ml的负载,以及6ml的本发明色谱膜的负载);3)用8mv洗涤缓冲液以10mv/min洗涤;4)用10mv以5mv/min洗脱;以及5)用5mv洗涤缓冲液以10mv/min洗涤。然后通过平衡开始下一个循环(步骤1),或者在重新开始之前,使用10mv的0.1m氢氧化钠以3mv/min进行原位清洗(cip)步骤,这导致一个cip步骤的持续时间为3分钟,接着使用平衡缓冲液以10mv/min的流速进行另外的洗涤步骤,直到达到ph7.4。
[0183]
cip步骤的频率随着循环实验的进行而减小,以降低cip步骤的清洁强度。对于前20个循环,在每个循环之后进行cip步骤。对于循环数21-40,cip步骤仅在每5个循环之后进行。对于循环数41-70,cip步骤仅在每10个循环之后进行。对于从第71个循环开始的所有更多循环,不再进行cip步骤。
[0184]
图11显示了两个装置在10mv/min的流速下的平衡阶段期间在每个循环中观察到的反式装置(δ)压力。
[0185]
首先,重要的是认识到,尽管两种材料具有非常相似的配置(对于两种材料床高度都是约1mm),但在10mv/min的相同流速下,与本发明的色谱膜的压力(0.01mpa)相比,对流材料的反式装置压力(约0.07mpa)要高得多。这是由于对于来自cytiva的对流纤维材料所观察到的较小的孔径导致的,这是获得与本发明的色谱膜的结合能力相似的结合能力的先决条件。
[0186]
来自cytiva的对流纤维材料在前20个循环期间显示出稳定的反式装置压力,在每个结合和洗脱循环之后是cip步骤。然而,由于cip频率在第21个循环之后降低到仅在每5个循环之后进行一次cip步骤,所以纤维材料开始表现出快速增加的反式装置压力。在第31个循环中,反式装置压力达到0.76mpa,这在这种材料的相关工业应用中很好地超过了高达0.4mpa的典型加工压力。
[0187]
本发明的色谱膜样品a在连续的循环中显示出关于反式装置压力发展的不同行为:反式装置压力在几乎79个循环中保持在0.01mpa的低水平,尽管cip频率在79个循环期间持续降低,直到在第70个循环之后不再进行cip步骤的事实。在第79个循环之后开始,观察到反式装置压力缓慢增加,在第150循环下达到约0.47mpa。尽管事实是结合能力在相同水平上,但这两种材料的这种行为明显不同。
[0188]
数据清楚地显示了两种分离材料的明显不同的结垢倾向。与基于纯对流纤维素纤维的膜材料(约0.4μm)相比,这种行为很可能是由于本发明的色谱膜的显著更大的对流孔径(约4μm)。
[0189]
实施例10:整合到可扩展装置系列中
[0190]
本发明的色谱膜样品a(参见实施例8)使用连续涂覆方法以卷形式制造,其中将乳液涂覆到第三惰性相上。将乳液冷却至胶凝点以下以形成连续的膜卷。
[0191]
含有表面活性剂和溶剂的本发明的色谱膜卷材料通过使用醇和水的连续萃取和洗涤过程进行萃取。随后,卷料进行交联并且同时通过用含有氢氧化钠的双环氧乙烷溶液
浸渍而活化。在连续洗涤过程中洗涤后,蛋白质a的偶联在跳汰机(jigger)中通过在两个卷绕辊之间卷绕活化的膜卷来进行,从而使溶解在磷酸盐缓冲液(1m,ph8.5)中的含有蛋白质a(10mg/ml)的反应溶液通过。随后,在连续冲洗/干燥过程中,从湿润剂中干燥蛋白质a膜,得到卷形式的蛋白质a膜。
[0192]
从这些膜卷,可以产生在恒定床高度下具有三个不同床体积的螺旋卷绕色谱模块。通过改变堆叠的本发明色谱膜的层数,可以基于目标值如渗透率、结合能力或轴向色散来建立各种床体积和床尺寸。在这些模块中,膜以交替的方式与间隔物羊毛卷在一起。
[0193]
通过熔融工艺以流体密封的方式将膜层连接到外壳。膜床的边缘在整合到相应的外壳中之前在前一步骤中被密封。从膜床到入口的上游给出冲击流通道,并且在出口的下游给出流出通道。
[0194]
表5示出了床体积从1.2至70ml不同的三个模块的特性,这对应于58.3的比例因子。床高度保持恒定在4mm,确保对于7.6-8.3mv/min*巴的所有三个模块几乎相同的渗透率,尽管床体积不同。
[0195]
使用现有技术的色谱系统,如实施例9中所述,用三个模块中的每一个运行澄清mab进料流的单一色谱结合和洗脱循环,以便评价色谱性能的相似性水平。对于1.2ml纳米装置,使用如实施例8中所述的150 avan系统。对于10ml微胶囊和70ml 5-英寸胶囊,使用多用途膜色谱系统(sartorius,mcsceb46nak)。所述系统是旨在用于生产和工艺开发的液相色谱系统,由基于快速循环色谱(rcc)的膜色谱实施配置组成。
[0196]
图12中描绘了归一化到透过各个模块的膜体积(x轴)的所产生的各个结合和洗脱循环的uv色谱图。三个装置的色谱图几乎是相同的,这是可扩展性能的强指示,即,尽管模块尺寸的范围很宽,即所使用的床体积的范围很宽,但色谱性能非常相似。
[0197]
表5:所使用的色谱模块的特征
[0198] 纳米胶囊微胶囊5英寸胶囊床体积[ml]1.21070床高度[mm]444渗透率[mv/min*巴]7.68.18.3洗脱体积[ml]5.05.35.3
[0199]
实施例11:不同进料流的稳健循环行为
[0200]
为了证明本发明的色谱膜的通用性和广泛的适用性,在快速循环过程中,用根据实施例6产生的增强的本发明的色谱膜纯化含有三种不同mab的三种不同的澄清mab发酵液,具有根据实施例7所述的第三惰性相a的样品编号34根据实施例1所述进行改性。如实施例10中所述,将本发明的色谱膜整合到具有1.2ml床体积的小规模装置中。对于具有200个循环的每个循环研究,使用新的装置。
[0201]
表6给出了对三种不同单克隆抗体进行的循环研究的概述。平衡后,给装置装载进料至先前测定的dbc 10%值的80%,然后洗涤、mab的洗脱、cip循环和再平衡。在表6的第3栏中,示出了用于不同步骤的所施加的体积。对于cip循环,体积是可变的,因为装置用0.2m naoh冲洗,直到装置流出物的ph达到ph 12.5,并在达到该ph值后再冲洗3.3mv。对于再平衡步骤,施加的pbs缓冲液体积也是可变的,随着装置被冲洗直到流出物达到7.5的ph。在所有循环研究期间,通过使用连接在样品泵和注射阀之间的sartopore2 xlg 0.8/0.2μm
(5441307g4
‑‑
ss,210cm2)进行进料的在线预过滤。
[0202]
在avant 150色谱系统下循环:
[0203]
表6:循环方法
[0204][0205]
图13、14和15示出了作为渗透体积的函数的每种抗体的各自200个循环的叠加uv色谱图。在所有200个循环中,所有三个色谱图都显示出非常相似和一致的行为,表明本发明的色谱膜在循环研究过程中没有改变其特性或性能。这也可以从图16、17和18中看出,图16、17和18分别示出了每种抗体/循环研究的各自200个循环的叠加的反式装置压力。在所有200个循环中,所有三个压力迹线都显示出非常相似和一致的行为,再次表明本发明的色谱膜在循环研究过程中没有改变其特性或性能。在200个循环的过程中可以观察到压力的轻微降低。这种反式装置压力的降低可以归因于在研究期间进料溶液从扩展研究开始时的4℃缓慢加热至室温,从而降低进料溶液的粘度并因此产生降低的反式装置压力。
[0206]
从100mau(第一次信号超过该阈值)-100mau(信号降至该阈值以下)分馏洗脱峰(uv色谱图中的第二个峰,从30-35ml体积开始),并测定产率、洗脱体积、洗脱浓度、污染物去除(hcp、dna、聚集体)以及浸出的proa配体的水平。用于检测产率、洗脱浓度和聚集体的方法是使用来自phenomenex的yarra sec 3000柱,使用thermo fisher scientific的hplc系统的尺寸排阻色谱法。用hcp elisa cygnus技术f#550第3代进行hcp分析。通过使用picogreen测定p11496分析dna。用repligen 9000/1的elisa检测洗脱级分中的proa配体浓度,然而,仅针对mab a洗脱级分。
[0207]
图19至25示出了mab a、mab b和mab c的数据集。很明显,所有三种mab的所有相关工艺特性(产率、hcp去除、dna去除、proa配体浸出、洗脱峰宽、池浓度、聚集物浓度)在200次循环过程中都保持在恒定水平。这证实了对于不同的mab和相应的进料流,所有三个装置的稳定的性能行为,尽管事实是仅使用了一个通用的循环配方。这些结果显示了本发明的色谱膜的非常稳健和进料流非依赖性的行为。
[0208]
实施例12:由于高床高度导致的高色谱性能
[0209]
根据实施例6产生增强的本发明的色谱膜,具有根据实施例7所述的第三惰性相b的样品编号34根据实施例1所述进行改性。为了证明床高度对色谱性能的影响,通过将不同
数目的膜层(即3层和12层)堆叠到装置中,将所生成的本发明色谱膜构建成两种不同床高的色谱装置。在色谱床之前以及在色谱床之后的流动分布对于两个装置都是相同的,与床高度无关。区别仅在于整合的本发明的色谱膜层数的这两个色谱装置具有以下特征:
[0210]
表7
[0211] 床高度反式装置压力以5mv/min下降3层0.78mm0.007mpa12层3.1mm0.043mpa
[0212]
图26和27中的色谱图示出了仅在床高度上不同的装置的不同色谱行为。突破曲线和洗脱峰都显示出了明显的差异。对于仅具有3层本发明的色谱膜的低床高度装置,可以观察到早期突破、突破曲线的缓慢增加和宽洗脱峰,而对于具有12层本发明的色谱膜的高床高度装置,观察到晚期突破、相对尖锐的突破增加和窄洗脱峰。这些影响导致较高的结合能力和较快的洗脱,导致较高的生产率、较低的缓冲液消耗和较高的洗脱池浓度。这些影响可归因于由于较低的轴向色散而导致的具有较高床高度的装置的改善的色谱行为。
[0213]
实施例13:用阳离子交换或混合模式配体官能化的本发明色谱膜的材料和性能特征。
[0214]
为了证明本发明色谱膜的通用性和平台特性,将不同的官能团固定到根据上述一般制备例中所述的实施例6产生的本发明色谱膜(样品编号34和35)上。
[0215]
ciex官能化:
[0216]
ciex配体官能化在两步法中进行。在第一步中,根据实施例6,本发明色谱膜(样品编号35)的活化用双环氧乙烷分子进行,使得双环氧乙烷的至少一个环氧乙烷基团与膜反应以增强基质材料的机械稳定性。第二步是通过在碱性ph下将配体添加到活化的本发明色谱膜上进行。对于该实施例,在24小时的时段内,将3-溴丙磺酸盐/酯(3-bps)在ph=14下以20wt%的反应溶液浓度固定在本发明的色谱膜上。表8列出了所产生的阳离子交换膜的性质。
[0217]
表8:
[0218][0219][0220]
为了表征根据实施例13产生的ciex官能化的本发明色谱膜(样品编号36)的结合和洗脱性能,在具有pp过滤器外壳(过滤台和过滤器盖)和不锈钢保持器的可重复使用的过滤装置中将膜组装成4层膜堆叠,如实施例8所述。
[0221]
根据实施例1中所述的方法,在不同流速下测定本发明的色谱膜的dbc 10%值,以获得目标蛋白γ-球蛋白(sigma-aldrich,g5009)的不同停留时间(rt)。在结合缓冲液条件
(乙酸钠,20mm,ph=5,1.5ms/cm)下,以15、10、7.5、5、2.5和1mv/min的流速,将目标蛋白γ-球蛋白(0.88g/ml)装载到本发明的色谱膜上,这导致4、6、8、12、24和60s的停留时间。在相同条件下检查sartobind s(sartorius目录,96iexs42euc11-a),代表现有技术的膜材料。
[0222]
作为代表现有技术的ciex树脂材料的基准材料,1ml hitrap capto柱(cytiva目录,29400458)购自树脂具有90μm的平均珠粒尺寸,代表扩散相。通过在相同的结合缓冲液条件下以5、1、0.5和0.2mv/min的流速(其等同于如上文针对膜材料所述的0.2、1、2和5分钟的停留时间)测定γ-球蛋白(4.74g/ml)的dbc 10%值来表征填充有capto树脂的柱的色谱性能。
[0223]
图28示出了作为ciex材料的停留时间的函数的dbc 10%结果。
[0224]
结果显示,与各自的现有技术的树脂相比,本发明的色谱ciex膜的dbc 10%增加了47%,与现有技术的膜相比,本发明的色谱ciex膜的dbc 10%增加了62%,同时分别使目标分子在色谱材料中的停留时间缩短90%或相同。
[0225]
混合模式(mm)官能化:
[0226]
在根据实施例6产生的本发明的色谱膜(样品编号34)上进行具有永久正电荷的混合模式配体的配体固定。根据上述一般制备例使用双环氧乙烷分子进行活化,然后用烯丙基溴(20wt%,2mol/lnaoh,24h)进行烯丙基化。根据us 8,895,710b2进行溴化和最终配体(n-苄基-n-甲基乙醇胺,bmea)偶联。
[0227]
根据实施例1,本发明的色谱mm膜关于配体密度、sbc和dbc10%进行表征。
[0228]
本发明的色谱mm膜的配体密度根据本领域技术人员已知的用hcl滴定ciex材料来测定。在结合缓冲液条件(甘氨酸-naoh,20mm,ph=8,40μs/cm)下,用γ-球蛋白(3mg/ml)作为目标测定sbc。洗涤后的洗脱步骤在洗脱条件缓冲液(甘氨酸-hcl,20mm,ph=2.5,2.1ms/cm)中进行。
[0229]
本发明的色谱mm膜的表征结果示于表9中。
[0230]
表9:
[0231][0232]
在该实施例中,根据实施例13的本发明的色谱膜(样品编号37)在具有pp过滤器壳体(过滤器台和过滤器盖)和不锈钢保持器的可重复使用的过滤装置中组装成膜堆叠,并根据实施例1中所述的方法进行表征。
[0233]
用γ-球蛋白(1mg/ml)作为目标和用于sbc测定的条件进行dbc 10%的测定。在各种流速下测定dbc 10%值以获得目标分子在色谱材料中的不同停留时间(rt)。将流速调节
至1、5、10和20mv/min,这对应于1.0、0.2、0.1和0.05min的停留时间。
[0234]
1ml hitrap capto adhere柱购自cytiva(cytiva目录,28405844),并用作代表现有技术的混合模式树脂材料的基准材料。树脂具有75μm的平均珠粒尺寸,代表扩散相。通过测定γ-球蛋白(sigma-aldrich,目录g5009)在2分钟和4分钟的停留时间(其相当于如上所述的ciex膜的2和4mv/min的流速)下的10% dbc值来表征这种现有技术的混合模式树脂材料的色谱性能。
[0235]
作为停留时间的函数的dbc 10%的结果描绘在图29中。结果显示,与相应的现有技术的mm树脂相比,本发明的色谱mm膜的dbc 10%增加了280%,同时目标分子在本发明的色谱mm膜中的停留时间缩短了95%。
[0236]
具有不同配体的性能数据显示了本发明的色谱膜的通用性,以及其对于不同色谱任务的适用性。由于多糖易于官能化,可以将多种官能配体固定到本发明的色谱膜上,产生相应的色谱材料。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1