硅胶及其制备方法与流程
1.本公开涉及硅胶吸附剂领域,尤其涉及一种用于purex流程中zr离子去除的硅胶及其制备方法。
背景技术:
2.在乏燃料后处理的钚铀还原提取(purex)流程和高放废液萃取分离等过程中,在槽油-水界面处,经常会形成含有固体的乳化层而妨碍工艺的正常运行,严重时甚至使萃取流程被迫停止。这个乳化层通常被称为“界面污物”,研究发现zr离子与萃取剂和稀释剂的降解产物间形成复杂的不溶性胶质体是界面污物产生的关键因素。因此,预先去除zr离子是切断界面污物生成途径、减缓界面污物形成的有效方式。
3.国内外针对purex流程中zr离子的去除开发了硅胶、金属氧化物、有机树脂、海藻酸钠凝胶等多种吸附剂(林灿生,裂变产物元素过程化学[m],中国原子能科学出版社,北京,2012,216-220;onishi t,et al.progress in nuclearenergy,2015,82:69-73)。其中,硅胶因其优异的辐照稳定性和化学稳定性而最有希望用于乏燃料后处理中。在模拟后处理工况下,梁俊福等研究了市售层析硅胶(比表面积330m2/g,平均孔径10.1nm)对zr的吸附,发现在3m hno3体系中层析硅胶对zr的最大吸附容量仅17.88mg/g,吸附平衡时间却长达50h 以上(梁俊福等,核化学与放射化学,2006,28:80-85)。ahrland等也报道了类似的结果(ahrland s.,et al.acta chemica scandinavica,1960,14:1059-1076)。这说明普通市售硅胶存在吸附容量低、平衡时间长等显著缺点。张裕卿等人报道了一种自制高比表面积的硅胶(比表面积998m2/g、平均孔径2.5nm),其对 zr离子的饱和吸附容量可达到32.6mg/g,但吸附平衡时间并未有效改善,仍长达48小时以上(张裕卿等,核化学与放射化学,2000,22:156-160)。
[0004]
使用这些吸附容量低、动力学缓慢的硅胶材料在实际工艺过程中需要频繁更换填料,不仅影响工艺的正常运行,还会产生大量难以处理的放射性固体废物。因此,急需开发purex流程中zr的高效吸附剂。
技术实现要素:
[0005]
有鉴于此,本公开提供一种制备硅胶的方法以及由此获得的硅胶,该硅胶具有良好的吸附容量,且具有显著改善的吸附平衡时间,极大地提升了purex 流程中zr的吸附效率。
[0006]
本公开的第一方面提供了一种硅胶的制备方法,包括用环糊精和季铵盐双子表面活性剂制备模板溶液,向所述模板溶液中加入有机硅氧烷,并进一步加入碱,得到固体产物,以及对所述固体产物焙烧制得所述硅胶,其中,所述环糊精选自由α-环糊精、β-环糊精、γ-环糊精中组成的组中的一种,且所述季铵盐型双子表面活性剂具有下式(1)所示结构,其中,r1~r4为甲基或乙基,n 为2~10的整数,优选的n为2~8的整数,r5和r6为碳原子数8~16之间的正烷基,优选的r5和r6为碳原子数12~16之间的正烷基,x-为cl-、br-或i-;
[0007][0008]
根据本公开的一种实施方式,所述环糊精和所述季铵盐型双子表面活性剂的摩尔比为1:(0.2~0.6),优选地为1:(0.35~0.55),且所述模板溶液中,所述环糊精和所述季铵盐型双子表面活性剂两者的质量浓度之和为2~15wt%,优选地为4~12wt%。
[0009]
根据本公开的一种实施方式,所述环糊精和所述有机硅氧烷的摩尔比为 1:(10~30),优选地为1:(15~25),所述有机硅氧烷为正硅酸酯,优选地为正硅酸甲酯、正硅酸乙酯、正硅酸丙酯、正硅酸丁酯或正硅酸四异丙酯。
[0010]
根据本公开的一种实施方式,所述碱包括水溶性有机碱和无机碱,且所述碱的加入量使加入后体系ph为9.0~13,优选地为10~12。
[0011]
根据本公开的一种实施方式,该碱选自nh3h2o、naoh、koh、(ch3)4noh 和(ch2ch3)4noh。
[0012]
根据本公开的一种实施方式,所述模板溶液的制备包括将溶解有所述环糊精和所述季铵盐双子表面活性剂的水溶液在20~80℃下搅拌0.5~1小时,以及在 20~40℃下静置4~12小时。
[0013]
根据本公开的一种实施方式,向所述模板溶液中加入所述有机硅氧烷后,搅拌5~20分钟,并在加入所述碱后,搅拌1~3小时,然后在20~80℃下静置 12~36小时。
[0014]
根据本公开的一种实施方式,所述焙烧在500~650℃下进行4~8小时。
[0015]
本公开的第二方面提供了一种硅胶,所述硅胶根据上述任一种制备方法制备得到。
[0016]
根据本公开的一种实施方式,所述硅胶在0~100nm范围内的孔径分布具有两个或两个以上的峰,其中面积最大的两个峰,分别在0~10nm和15~100nm 两个范围内,所述硅胶的比表面积为400m2/g~1000m2/g,所述硅胶的孔容为 0.6~1.2ml/g,所述硅胶对zr离子的饱和吸附容量可达到50mg/g以上,吸附平衡时间在6~8小时。
[0017]
本公开采用环糊精-季铵盐双子表面活性剂为混合模板剂制备硅胶同时获得两种以上不同孔径大小的多级孔结构,可用于purex流程中zr离子的选择性去除,吸附动力学高,对zr离子的吸附平衡时间在6~8小时,缩短到张裕卿等报道的硅胶的吸附平衡时间的1/8~1/6;吸附活性高,饱和吸附容量均在25 mg/g以上,甚至高达50mg/g以上,比张裕卿等报道的硅胶的饱和吸附容量提高了50%以上。
附图说明
[0018]
图1为实施例1所得的多级孔结构硅胶的扫描电镜图;
[0019]
图2为实施例1所得的多级孔结构硅胶的透射电镜图;
[0020]
图3为实施例2所得的多级孔结构硅胶的扫描电镜图;
[0021]
图4为实施例2所得的多级孔结构硅胶的透射电镜图;
[0022]
图5为实施例2所得的多级孔结构硅胶材料的氮气吸附-脱附等温线图和相应的孔径分布图;
[0023]
图6为实施例3所得的多级孔结构硅胶材料的氮气吸附-脱附等温线图和相应的孔
径分布图;
[0024]
图7为实施例5所得的多级孔结构硅胶的扫描电镜图;
[0025]
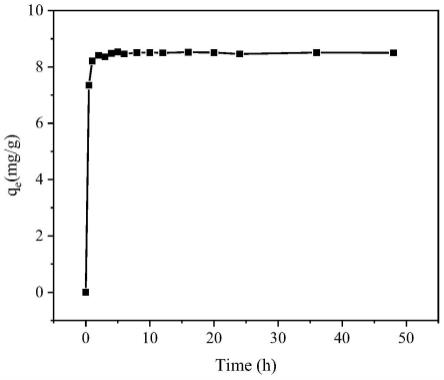
图8为实施例1所制备的多级孔结构硅胶对zr离子的吸附动力学曲线;
[0026]
图9为实施例2所制备的多级孔结构硅胶的用量对zr离子吸附的影响。
具体实施方式
[0027]
下面将结合本公开实施方式及附图,对本公开实施方式中的技术方案进行清楚、完整地描述,显然,所描述的实施方式仅仅是本公开的一部分实施方式,而不是全部的实施方式。基于本公开中的实施方式,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施方式,都属于本公开保护的范围。
[0028]
如上所述,本公开意在提供一种具有改进的吸附动力学和容量的、在 purex流程中高效吸附zr的硅胶。根据本公开的实施例,提供一种硅胶的制备方法,其特征在于,在使用环糊精和季铵盐双子表面活性剂配制模板溶液中,利用碱性条件水解硅源制备硅胶,其中,在所述模板溶液中,所述环糊精和所述季铵盐双子表面活性剂的摩尔比为1:(0.2~0.6)。
[0029]
季铵盐双子表面活性剂
[0030]
季铵盐双子表面活性剂的制备方法在本公开中没有特别限制,例如,可通过1-溴代长链烷烃和n,n,n’,n
’‑
四甲基烷基二胺在无水乙醇中加热回流,进行季铵化反应而制得。季铵盐双子表面活性剂具有独特的结构,两个亲水的季铵基和两个疏水的长链烷基通过联接基团连接,使得双子表面活性剂能够更有效地降低水的表面张力,易聚集生成胶束结构,有更低的临界胶束浓度。由于季铵盐双子表面活性剂结构可变性强,其中影响其特性的重要因素包括连接基的刚性、亲水性和尺寸等,通过改变联接基团的化学结构可方便地调控表面活性剂的胶束结构,进而影响以该胶束为模板的形成的多孔材料的结构。
[0031]
根据优选的实施方式,季铵盐型双子表面活性剂具有下式(1)所示结构,其中,r1~r4为甲基或乙基,n为2~10的整数,r5和r6为c8~16的直链烷基, x-为cl-、br-或i-;优选的n为2~8的整数,r5和r6为c12~16的直链烷基,
[0032][0033]
环糊精
[0034]
环糊精是直链淀粉在由芽孢杆菌产生的环糊精葡萄糖基转移酶作用下生成的一系列环状低聚糖的总称,通常有α-环糊精、β-环糊精,γ-环糊精3种,组成它们的吡喃葡萄糖分子数为6、7、8。环糊精在碱性介质中很稳定,但强酸可以使之水解,对酸的耐受性比直链淀粉强。环糊精分子具有略呈锥形的中空圆筒状结构,外侧上端(较大开口端)由c2和c3的仲羟基构成,下端(较小开口端)由c6的伯羟基构成,具有亲水性,内侧是空腔,空腔内是疏水区,可嵌入各种有机化合物,比如表面活性剂,形成复合物。α-环糊精、β-环糊精、γ-环糊精的空腔由小到大,在本公开中,都可以用来形成复合物。优选的,使用空腔适中且生产成本低的β-环糊精。
[0035]
环糊精与季铵盐双子表面活性剂的模板溶液
[0036]
环糊精在和表面活性剂形成聚集体的相互作用的过程中起可能调节剂和/ 或构建单元的作用。作为调节剂,尽管环糊精本身不参与最终的表面活性剂聚集体,但它们可以通过结合聚集体中的表面活性剂分子的方式极大地影响聚集体。作为结构单元,环糊精可以附着在疏水部分,或者环糊精/表面活性剂复合物组装成聚集体。
[0037]
由于复杂的作用方式与季铵盐双子表面活性剂的特殊结构,在环糊精/季铵盐双子表面活性剂的溶液中,可能存在多种复杂的聚集体形态作为后序硅胶形成的模板,取决于环糊精和季铵盐双子表面活性剂摩尔比。根据优选的实施方式,在本公开的模板溶液中,环糊精和所述季铵盐双子表面活性剂的摩尔比为 1:(0.2~0.6),优选地为1:(0.35~0.55),两者之和的质量浓度为2~15wt%,优选地为8~12wt%。出乎意料地,以该模板溶液制得的硅胶,对zr离子的吸附平衡时间可以控制到6~8小时,饱和吸附容量均在25mg/g以上,有的甚至高达50mg/g 以上。
[0038]
有机硅氧烷
[0039]
有机硅氧烷是形成硅胶的硅源。本公开的有机硅氧烷可选自正硅酸酯,实例包括正硅酸甲酯、正硅酸乙酯、正硅酸丙酯、正硅酸丁酯、正硅酸四异丙酯等。环糊精和有机硅氧烷的摩尔比为1:(10~30)。
[0040]
硅胶的制备方法
[0041]
硅胶的制备方法具体包括以下步骤:
[0042]
1)将环糊精和季铵盐双子表面活性剂分散在水中,搅拌,然后静置陈化,获得模板溶液;
[0043]
2)向所述模板溶液中加入有机硅氧烷,搅拌;
[0044]
3)向步骤2)所得的溶液中加入碱,搅拌并静置后,得到固体产物;
[0045]
4)将步骤3)的产物过滤、水洗、干燥;
[0046]
5)将步骤4)的产物焙烧后制得所述硅胶。
[0047]
本公开模板溶液的配制过程,涉及环糊精和季铵盐双子表面活性剂在水中溶解的过程,以及该表面活性剂聚集体和该环糊精与该表面活性剂结合体之间形成平衡的过程,需要选择条件,保证充分的溶解和平衡。本公开的步骤1)中在温度为20~80℃的条件下进行搅拌,时间为0.5~1小时,静置陈化的温度为 20~40℃,时间为4~12小时。
[0048]
本公开的方法的步骤2)中搅拌的时间为5~20min。
[0049]
酸性条件或碱性条件都可以催化有机硅氧烷水解和缩聚形成硅胶。有机硅氧烷在碱性条件下,其水解步骤主要涉及硅酸根和氢氧根离子对硅原子的进攻,速度比酸性条件更快,而缩聚后形成更加支化的结构。随后逐渐形成胶体颗粒,颗粒间堆积进一步形成网状的硅胶。本公开优选碱性条件下的制备硅胶。用于本公开方法的碱可选自nh3h2o、naoh、koh、(ch3)4noh、(ch2ch3)4noh 等常见的水溶性有机碱和无机碱。碱的加入量优选地确保将体系的ph值调为 9~13,优选地为10~12。步骤3)中搅拌的时间为1~3小时,步骤3)中所述静置反应的温度为20~80℃,时间为12~36小时。
[0050]
刚形成的硅胶样品主要以无定形二氧化硅的形式存在,随着加热温度的升高,600℃时已经能够除尽表面活性剂和一些杂质,并逐渐出现了结晶区,但当煅烧温度升到800℃以上时,开始出现了明显的熔结和塌陷,结构遭到了破坏。因此,本公开步骤5)中所述焙烧的温度为500~650℃,时间为4~8小时。
[0051]
在上述用量比例范围内,引入环糊精-双子表面活性剂为混合模板剂,在硅胶合成中,制备了一类新型多级孔结构硅胶,获得了预料不到的zr吸附的吸附动力学效果和改进的吸附容量。通常把孔径划分为以下3类,微孔尺寸2~5nm,介孔尺寸20~30nm,大孔尺寸50~100nm。本公开的硅胶既含有微孔,又含有介孔和大孔。通过多级孔结构的引入,一方面利用大量的微孔结构提高硅胶的比表面积,进而增加吸附位点数量和提升材料的吸附容量;另一方面,硅胶内部大孔、介孔、微孔的相互贯通的多级孔道网络会使分子/离子在其内部具有更快的传输和扩散速率,大幅提高材料的传质性能,进而有效缩短吸附平衡时间。此外,由该特殊膜板剂形成的多级孔结构,还可能提高了表面缺陷和表面不饱和原子的比例,也可能造成了特殊的形状的孔,显著增加材料对zr的吸附活性。所得硅胶的比表面积达到400m2/g~1000m2/g,孔容达到0.6~1.2ml/g。
[0052]
以下通过具体实施例来进一步说明本公开的技术方案。
[0053]
实施例1
[0054]
将0.757g的β-环糊精和0.136g的丙撑基(十六烷基二甲基溴化铵)加入19.1 ml水中,在60℃下快速搅拌0.5h,然后在35℃下陈化12h,获得模板溶液。在快速搅拌条件下,向模板溶液中加入3ml正硅酸乙酯,继续搅拌5min,并在快速搅拌条件下,进一步加入0.3ml nh3h2o溶液(浓度28wt%),体系ph 为9,继续搅拌120min,然后在35℃下静置反应24小时。将所得的固体产物过滤,并用水洗3次,然后将固体产物在80℃下干燥12h,然后置于马弗炉中,以1℃/min升温速率升至550℃,然后在550℃焙烧5h,得到多级孔结构硅胶,其形貌为片状(如图1和图2所示),测得比表面积为597.2m2/g,具有双孔分布(孔径1为约2.8nm,孔径2为约20.5nm),孔容为0.78ml/g。
[0055]
实施例2
[0056]
将1.30g的α-环糊精和0.350g的乙撑基(十二烷基二甲基氯化铵)加18.25 ml水中,在80℃下快速搅拌1h,然后在40℃下陈化6h,获得模板溶液。在快速搅拌条件下,向模板溶液中加入6ml正硅酸乙酯,继续搅拌10min,并在快速搅拌条件下,进一步加入1.2ml nh3h2o溶液(浓度28wt%),体系ph 为10,继续搅拌120min,然后在80℃下静置反应24小时。其余步骤同实施例 1,所得产物形貌为不规则形状(如图3和图4所示),测得比表面积为976.2m2/g,双孔分布(孔径1为~2.8nm,孔径2为~55nm,如图5所示),孔容为1.131ml/g。
[0057]
实施例3
[0058]
将1.72g的β-环糊精和0.273g的辛撑基(十六烷基二甲基氯化铵)加入18 ml水中,在60℃下快速搅拌1h,然后在40℃下陈化6h,获得模板溶液。在快速搅拌条件下,向模板溶液中加入6ml正硅酸乙酯,继续搅拌10min,并在快速搅拌条件下,进一步加入0.6ml nh3h2o(浓度28wt%),体系ph为9,继续搅拌120min,然后在60℃下静置反应32小时。其余步骤同实施例1,所得产物形貌为不规则形状,测得比表面积759.6m2/g,具有双孔分布(孔径1 为~2.8nm,孔径2为~30nm,如图6所示),孔容为0.673ml/g。
[0059]
实施例4
[0060]
按照实施例3的方法,区别是将辛撑基(十六烷基二甲基氯化铵)的用量由 0.273g增加为0.546g,将ph调为12。所得产物形貌为不规则形状,测得比表面积为944.5m2/g,具有双孔分布(孔径1为~2.8nm,孔径2为~25nm),孔容为0.97ml/g。
[0061]
实施例5
[0062]
按照实施例1的方法,将步骤(1)中0.757g的β-环糊精变为0.865g 的γ-环糊精,其余条件不变,所得产物形貌为不规则形状(如图7所示),测得比表面积为405.5m2/g,具有双孔分布(孔径1为~2.8nm,孔径2为~30nm),孔容为0.80ml/g。
[0063]
图1到图7表明,通过改变环糊精-表面活性剂的种类和比例、反应温度等条件,可以使得产物的尺寸、孔径大小及分布、比表面积和孔容等关键参数发生显著的改变,从而可对硅胶吸附性能产生影响。
[0064]
测试例1
[0065]
分别称取15份50mg实施例1制得的硅胶,分别加入到5ml的zr(no3)4的hno3溶液中(其中,hno3浓度为3m,初始zr离子浓度100mg/l),25℃下搅拌,然后分别在0.5、1、2、3、4、5、6、8、10、12、16、20、24、36、 48h等时间点取上清液,过滤后用icp-aes测试zr离子浓度,作图以获得多级孔结构硅胶对zr离子的吸附平衡时间(如图8所示)。利用同样的方法研究了实施例2~5所得的硅胶,其结果如表1所示。
[0066]
测试例2
[0067]
称50mg实施例1制得的硅胶,加入到5ml的zr(no3)4的hno3溶液中, hno3浓度为3m,初始zr离子浓度在10~500mg/l之间,25℃下搅拌12h,取上清液过滤后测定其浓度,得到zr离子浓度对吸附的影响(如图9所示)。并利用langmuir吸附等温线拟合得到实施例1制得的硅胶对zr离子的饱和吸附容量。利用同样的方法研究了实施例2~5所得的硅胶,其结果如表1所示。
[0068]
表1:实施例1~5的硅胶对zr离子的吸附性能
[0069] 平衡时间/h最大吸附容量mg/g实施例1631.4实施例2650.3实施例3638.6实施例4648.7实施例5826.9
[0070]
表1数据表明,本公开的硅胶,吸附动力学高,对zr离子的吸附平衡时间在6~8小时。对比张裕卿等人报道的硅胶(张裕卿等,核化学与放射化学,2000, 22:156-160),其zr离子的饱和吸附容量可达到32.6mg/g,吸附平衡时间长达 48小时以上,本公开制得的硅胶对zr离子的吸附平衡时间缩短到1/8~1/6,饱和吸附容量均在25mg/g以上,甚至高达50mg/g以上。
[0071]
测试例3
[0072]
称取100mg实施例2制得的多级孔结构硅胶,加入到10ml酸浓度为3m hno3的模拟放射性废液中(其组成如表2所示),30℃下搅拌12h,取上清液过滤后测试测定各离子的浓度,计算得到不同离子的去除率和分配系数,结果如表2所示。
[0073]
表2:实施例2的硅胶对模拟放射性废液各组分的去除效果
[0074][0075]
表2数据表明,本公开的硅胶,对zr离子的吸附有较高的选择性,对模拟放射性废液中其他组分的吸附性低。对比张裕卿等人报道的硅胶(张裕卿等,核科学与工程,2000,20(4):353),同样在3m的hno3环境下,对zr
4+
分配系数为56ml/g,仅为本公开实施例2的硅胶的66%。
[0076]
以上所述仅为本公开的优选实施方式,并非因此限制本公开的专利范围,凡是在本公开的公开构思下,利用本公开说明书及附图内容所作的等效结构变换,或直接/间接运用在其他相关的技术领域均包括在本公开的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1