一种适应中高浓度CO2高效捕集的膜分离技术的制作方法
一种适应中高浓度co2高效捕集的膜分离技术
技术领域
1.本发明属于高分子化学领域,更具体地为一种适应中高浓度co2高效捕集的膜分离技术。微孔材料由于自身刚性分子链的扭曲折叠等等会产生高比表面积的微孔结构,相应的膜材料具有优异的气体分离性能,将刚性扭曲的结构单元引入到聚酰亚胺主链中就得到自具微孔的聚酰亚胺。尤其涉及一种包含刚性扭曲单元的聚酰胺酸单体制备以及高分子聚酰亚胺的合成,用于co2高效捕集的分离膜。
背景技术:
2.关于气候快速变化的报道强调了生态系统的风险和对人类生活的严重影响。人为温室气体在大气中的不断积累是导致气候快速变化主要原因之一。人为温室气体主要包括ch4、no
x
和co2(后者约占人为温室气体的77%)。根据keeling曲线1.88亿吨的co2排放,将会导致大气中co2含量升高1ppm。ippc的报告预测,到2035年,co2浓度将升至450ppm,全球气温有可能上升2℃。2018年10月,ipcc发布的报告指出,与全球升温2℃相比,将全球变暖控制在1.5℃会更有利于人类和自然生态系统。进一步控制温度升高意味着我们会面对更大的挑战,同时也需要采取更多有效的措施。中国对世界承诺,与2005年相比,2030年单位国内生产总值co2排放量下降60%-65%co2排放量达到峰值并争取早日实现。co2的主要排放者是能源部门、钢铁和水泥工业,这些部门现有的缓解政策主要集中于提高能源效率过程、转向低碳密集能源、以及碳捕集和封存(ccs)技术。为了控制全球温度上升,实现co2的减排,大力开发和利用ccs技术成为一种必定的趋势。
3.co2捕集技术是指将co2从电厂、钢铁厂、煤化工厂等排放的烟气中分离出来,并避免co2直接排放到大气的一种技术。根据反应原理不同,co2捕集技术主要包括吸附法、吸收法、膜法和深冷分离法。根据co2的分压、工作条件和混合气体的组成,可以根据不同的来源,选择不同的技术捕获co2。
4.在co2捕集技术中,膜分离法是最有前途的环保方法之一。膜分离过程是以膜两侧的压力差异作为驱动力的压力驱动过程。膜分离捕集co2的原理是利用不同气体的扩散率、溶解度和吸附能力的差异将co2从混合气体中分离出来,因此在利用膜分离法捕集co2时不会发生物质相态变化。膜分离法的关键是膜材料的选择。根据膜的制备材料不同,可分为有机膜、无机膜和共混膜。目前研究较多的有机材料为聚酰亚胺(pi膜)。与传统co2捕集技术相比,膜分离法因分离性能高、成本低、环境友好等优点受到国内外研究者的广泛关注。但膜分离在工业上受到膜面积、寿命等条件限制,因此膜分离技术主要用于处理中小气量的co2捕集,在大型工艺中的应用还需要进一步提高膜的热稳定性、机械和化学稳定性。膜材料是膜分离法的关键。
5.为了提高中高浓度co2高效捕集的膜分离效率,本发明从分子结构角度设计了pi薄膜,该薄膜由六氟二酐和特殊制备的n
1-(1,1:4',1
”‑
三苯基]-3-基)-n
1-(4-氨基苯基)苯-1,4-二胺单体聚合而成,本结构中的-cf3基团通过强的吸电子效应提高链段刚性,从而提高了该材料的稳定性,大体积苯基侧基通过增大空间位阻降低了堆积密度,-cf3基团和
苯环侧基具有协同作用,共同增加了pi薄膜的自由体积,提高了膜的气体渗透性能、热稳定性、化学稳定性,从而实现co2高效捕集的膜分离技术。
技术实现要素:
6.气候变暖的形势越发严峻,实行co2减排措施刻不容缓。ccs技术成为最有效果的措施之一。目前co2捕集技术研究最多的包括吸附法、吸收法、膜分离法。膜分离法与其他捕集技术相比,具有分离性能高、成本低、对环境污染小等优点,有广阔的发展前景。膜的制备材料是膜分离法的关键。聚合物膜由于成本低廉、制备方法简单被广泛研宄,但其渗透性能和选择性互相制约,难以同时提高,限制了聚合物膜的工业应用。
7.为了提高co2高效捕集膜的分离技术,本发明提供如下技术方案:
8.s1、3-溴-n,n-双(4-硝基苯)苯胺的合成(n-tmbr):取用3-8g的3-溴苯胺配制为30mmol的溶液,再取用9-11g氟化铯配制为60mmol的溶液,加入500ml三颈圆底烧瓶中加热50-120℃进行混合溶解,时间约为1-2h。之后配制5-12g 1-氟-4-硝基苯浓度为75mmol,二甲基亚砜150-300ml、乙酸乙酯120-220ml继续添加到500ml三颈圆底烧瓶中,并在150℃-180℃氮气下反应24-48h。之后倒入500-900ml冷饱和盐水中,黄色沉淀物通过硅胶色谱法进行收集和纯化,并用二氯甲烷/己烷作为洗脱液,纯化后的产品为浅黄色针状晶体。
9.s2、n、n-双(4-硝基苯基)-[1,1':4',1
”‑
三苯基]-3-胺的合成(n-tmbp):取用s1步骤的产物2-8g n-tmbr,配制为10-15mmol和使用2-4g 4-联苯硼酸浓度配制为11-16mmol添加到500ml三颈圆底烧瓶,加热40-50℃用索氏提取器进行冷凝回流。之后取用0.2-0.5g用50-70ml乙醇溶液溶解三苯基膦钯,并加入35-45ml k2co3水溶液与200-500ml四氢呋喃,最后加入在氮气下回流20-72h。去除水层后,通过旋转蒸发和蒸发收集黄色沉淀物硅胶,并用二氯甲烷层析纯化/己烷作为洗脱液。纯化产物为浅黄色针状晶体。该步骤使用的索氏提取器为四联阀门款型号为jc-sstq2。
[0010]
s3、n
1-(1,1:4',1
”‑
三苯基]-3-基)-n
1-(4-氨基苯基)苯-1,4-二胺的合成(a-tmbp):取用步骤s2中得到的产物4-8g n-tmbp用乙酸乙酯配制成10-15mmol的溶液,并加入一匙0.05-0.08g 10%的钯/碳催化剂和100-400ml无水乙醇装入500ml三颈圆底烧瓶,然后,用滴液漏斗逐滴添加3-5ml水合肼并在氮气下回流24-48小时。移除通过旋转蒸发乙醇,收集灰色沉淀物并然后用二氯甲烷在硅胶上层析纯化/己烷作为洗脱液。纯化产物为灰色晶体。
[0011]
s4、pi薄膜(tmbphf)的合成与制备:取用0.3-0.7g s3步骤的a-tmbp用乙酸乙酯溶解为1-3mmol的溶液,取用0.3-0.8g六氟二酐溶于乙醇使其配成1-3mmol溶液,并纯化将5.8-6.9ml二甲基甲酰胺添加到50ml烧瓶中,混合物是室温下在氩气下搅拌约4-7小时,形成粘性5-(2-(4-(1,1:4',1
”‑
三苯基)-3-基(4-氨基苯基)氨基)苯基)异吲哚-5-基)-1,1,1,3,3,3-六氟丙烷-2-基)异吲哚-1,3-二酮溶液(tmbphf)。随后在干净干燥的玻璃板上均匀涂上控制薄膜厚度,然后在真空中进行热亚胺化温度程序为100℃-200℃的烘箱,以生产pi薄膜。tmbphf膜冷却至室温后从玻璃基板上移除。
[0012]
本发明中s2采用suzuki偶联反应,suzuki-miyaura反应(铃木-宫浦反应)零价钯配合物催化下,芳基或烯基硼酸或硼酸酯与氯、溴、碘代芳烃或烯烃发生交叉偶联。
[0013]
优选地:本发明中所选用的3-溴苯胺为纯度为96%以上的有机试剂。
[0014]
优选地:本发明所述的凝胶色谱柱的洗脱液全部为二氯甲烷层析纯化/己烷作为流动相。
附图说明
[0015]
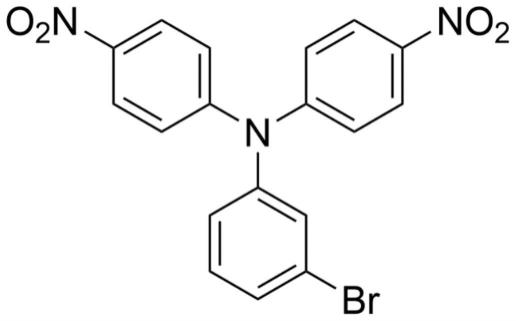
图1为本发明实施例1中n-tmbr有机化合物结构简式。
[0016]
图2为本发明实施例1中n-tmbr有机化合物核磁共振氢谱图。
[0017]
图3为本发明实施例2中n-tmbp有机化合物结构简式。
[0018]
图4为本发明实施例2中n-tmbp有机化合物核磁共振氢谱图。
[0019]
图5为本发明实施例3中a-tmbp有机化合物结构简式。
[0020]
图6为本发明实施例3中a-tmbp有机化合物核磁共振氢谱图。
[0021]
图7为本发明实施1与对比例1、对比例2中的co2选择性百分比柱状图。
[0022]
图8为本发明实施2与对比例3、对比例4中的co2选择性百分比柱状图。
[0023]
图9为本发明实施3与对比例5、对比例6中的co2选择性百分比柱状图。
[0024]
图10为本发明实施例4中tmbphf单体(pi薄膜单体)有机化合物结构简式。
[0025]
图11为本发明实施例4中tmbphf单体(pi薄膜单体)有机化合物核磁共振氢谱图。
具体实施方式
[0026]
气体分离膜是一种“绿色技术”利用混合气体中不同组分在压差的驱动下透过膜的速率不同而实现气体分离。膜分离具有分离效率高、耗能低、操作简单等优点,在与传统分离技术(吸附、吸收、深冷分离等)的竞争中显示出独特的优势,在气体的净化、纯化以及能源利用和环境治理中都有重要的作用,21世纪气体分离膜技术将取得长足发展,进而取代现有的吸收、萃取、精馏等耗能分离过程。气体分离膜的核心在于开发高通量、高选择性以及热稳定性、化学稳定性等更为理想的新型膜材料。
[0027]
实施例1
[0028]
s1、3-溴-n,n-双(4-硝基苯)苯胺的合成(n-tmbr):取用3g的3-溴苯胺配制为30mmol的溶液,再取用9g氟化铯配制为60mmol的溶液,加入500ml三颈圆底烧瓶中加热50℃进行混合溶解,时间约为1h。之后配制5g 1-氟-4-硝基苯浓度为75mmol,二甲基亚砜150ml、乙酸乙酯120ml继续添加到500ml三颈圆底烧瓶中,并在150℃氮气下反应24h。之后倒入500ml冷饱和盐水中,黄色沉淀物通过硅胶色谱法进行收集和纯化,并用二氯甲烷/己烷作为洗脱液,纯化后的产品为浅黄色针状晶体。
[0029]
s2、n、n-双(4-硝基苯基)-[1,1':4',1
”‑
三苯基]-3-胺的合成(n-tmbp):取用s1步骤的产物2g n-tmbr,配制为10mmol和使用2g 4-联苯硼酸浓度配制为11mmol添加到500ml三颈圆底烧瓶,加热40℃用索氏提取器进行冷凝回流。之后取用0.2g用50ml乙醇溶液溶解三苯基膦钯,并加入35ml k2co3水溶液与200ml四氢呋喃,最后加入在氮气下回流20h。去除水层后,通过旋转蒸发和蒸发收集黄色沉淀物硅胶,并用二氯甲烷层析纯化/己烷作为洗脱液。纯化产物为浅黄色针状晶体。
[0030]
s3、n
1-(1,1:4',1
”‑
三苯基]-3-基)-n
1-(4-氨基苯基)苯-1,4-二胺的合成(a-tmbp):取用步骤s2中得到的产物4g n-tmbp用乙酸乙酯配制成10mmol的溶液,并加入一匙0.05g 10%的钯/碳催化剂和100ml无水乙醇装入500ml三颈圆底烧瓶,然后,用滴液漏斗逐
滴添加3ml水合肼并在氮气下回流24小时。移除通过旋转蒸发乙醇,收集灰色沉淀物并然后用二氯甲烷在硅胶上层析纯化/己烷作为洗脱液。纯化产物为灰色晶体。
[0031]
s4、pi薄膜(tmbphf)的合成与制备:取用0.3g s3步骤的a-tmbp用乙酸乙酯溶解为1mmol的溶液,取用0.3g六氟二酐溶于乙醇使其配成1mmol溶液,并纯化将5.8ml二甲基甲酰胺添加到50ml烧瓶中,混合物是室温下在氩气下搅拌约4小时,形成粘性5-(2-(4-(1,1:4',1
”‑
三苯基)-3-基(4-氨基苯基)氨基)苯基)异吲哚-5-基)-1,1,1,3,3,3-六氟丙烷-2-基)异吲哚-1,3-二酮溶液(tmbphf)。随后在干净干燥的玻璃板上均匀涂上控制薄膜厚度,然后在真空中进行热亚胺化温度程序为100℃的烘箱,以生产pi薄膜。tmbphf膜冷却至室温后从玻璃基板上移除。
[0032]
对比例1除将步骤s1中的3-溴苯胺换为4-溴苯胺,其各步骤均与实施例1相同。
[0033]
对比例2除将步骤s1中的3-溴苯胺换为2-溴苯胺,其各步骤均与实施例1相同。
[0034]
表1
[0035]
检测项目对比例1对比例2实施例1co2选择性(%)60
±
0.0350
±
0.270
±
0.02
[0036]
图1为本发明实施例1中n-tmbr有机化合物结构简式,图2为本发明实施例1中n-tmbr有机化合物核磁共振氢谱图。图7为本发明实施例1和对比例1、2所得到的co2选择性百分比柱状图。表1为本发明实施例1和对比例1、2co2百分比平均数的统计表格。从图1和表1中可以看出实施例1中co2选择性百分比占比含量比较大,远高于对比例1、2。该结果表明,苯环间位上进行修饰刚性联苯,这种扭曲单元可以降低trade-off关系,3-溴苯胺为含氮芳杂环聚合物的间位排布,兼具优异的渗透性和选择性,是作为气体分离膜材料的最佳选择,其中,尤以聚酰亚胺的综合性能最佳,用来增加其co2高效捕集与分离效果。
[0037]
实施例2
[0038]
s1、3-溴-n,n-双(4-硝基苯)苯胺的合成(n-tmbr):取用8g的3-溴苯胺配制为30mmol的溶液,再取用11g氟化铯配制为60mmol的溶液,加入500ml三颈圆底烧瓶中加热120℃进行混合溶解,时间约为2h。之后配制12g 1-氟-4-硝基苯浓度为75mmol,二甲基亚砜300ml、乙酸乙酯220ml继续添加到500ml三颈圆底烧瓶中,并在180℃氮气下反应48h。之后倒入900ml冷饱和盐水中,黄色沉淀物通过硅胶色谱法进行收集和纯化,并用二氯甲烷/己烷作为洗脱液,纯化后的产品为浅黄色针状晶体。
[0039]
s2、n、n-双(4-硝基苯基)-[1,1':4',1
”‑
三苯基]-3-胺的合成(n-tmbp):取用s1步骤的产物8g n-tmbr,配制为15mmol和使用4g 4-联苯硼酸浓度配制为16mmol添加到500ml三颈圆底烧瓶,加热50℃用索氏提取器进行冷凝回流。之后取用0.5g用70ml乙醇溶液溶解三苯基膦钯,并加入45ml k2co3水溶液与500ml四氢呋喃,最后加入在氮气下回流72h。去除水层后,通过旋转蒸发和蒸发收集黄色沉淀物硅胶,并用二氯甲烷层析纯化/己烷作为洗脱液。纯化产物为浅黄色针状晶体。
[0040]
s3、n
1-(1,1:4',1
”‑
三苯基]-3-基)-n
1-(4-氨基苯基)苯-1,4-二胺的合成(a-tmbp):取用步骤s2中得到的产物8g n-tmbp用乙酸乙酯配制成15mmol的溶液,并加入一匙0.08g 10%的钯/碳催化剂和400ml无水乙醇装入500ml三颈圆底烧瓶,然后,用滴液漏斗逐滴添加5ml水合肼并在氮气下回流48小时。移除通过旋转蒸发乙醇,收集灰色沉淀物并然后用二氯甲烷在硅胶上层析纯化/己烷作为洗脱液。纯化产物为灰色晶体。
[0041]
s4、pi薄膜(tmbphf)的合成与制备:取用0.7g s3步骤的a-tmbp用乙酸乙酯溶解为3mmol的溶液,取用0.8g六氟二酐溶于乙醇使其配成3mmol溶液,并纯化将6.9ml二甲基甲酰胺添加到50ml烧瓶中,混合物是室温下在氩气下搅拌约4-7小时,形成粘性5-(2-(4-(1,1:4',1
”‑
三苯基)-3-基(4-氨基苯基)氨基)苯基)异吲哚-5-基)-1,1,1,3,3,3-六氟丙烷-2-基)异吲哚-1,3-二酮溶液(tmbphf)。随后在干净干燥的玻璃板上均匀涂上控制薄膜厚度,然后在真空中进行热亚胺化温度程序为200℃的烘箱,以生产pi薄膜。tmbphf膜冷却至室温后从玻璃基板上移除。
[0042]
对比例3除步骤s4中不加入六氟修饰的二酐外,将-cf3换为-ch3其余各步骤均与实施例2相同。
[0043]
对比例4除步骤s4中不加入六氟修饰的二酐外,将-cf3换为-c2h5其余各步骤均与实施例2相同。
[0044]
表2
[0045]
检测项目对比例3对比例4实施例2co2选择性(%)20
±
0.0640
±
0.0280
±
0.01
[0046]
图3为本发明实施例2中n-tmbp有机化合物结构简式,图4为本发明实施例2中n-tmbp有机化合物核磁共振氢谱图。图8为本发明实施例2和对比例3、4所得到的co2选择性百分比柱状图。表2为本发明实施例2和对比例3、4co2选择性百分比平均数的统计表格。从图8和表2中可以看出实施例2中co2选择性以及捕集分离效果较强,高于对比例3、4。该结果表明,聚酰亚胺中引入具有较高电负性和较低摩尔极化率的氟原子,可赋予了材料较高的透光率、较低的吸水率和介电常数。对气体分离膜而言,含氟基团的引入降低聚酰亚胺分子间的作用力,限制分子链的紧密堆积,增加自由体积,提高气体渗透性能,同时,较大的自由体积使溶剂分子易进入聚合物内部,从而提高聚酰亚胺的溶解性能,改善了加工性能,用来增加其co2高效捕集与分离效果。
[0047]
实施例3
[0048]
s1、3-溴-n,n-双(4-硝基苯)苯胺的合成(n-tmbr):取用4g的3-溴苯胺配制为30mmol的溶液,再取用10g氟化铯配制为60mmol的溶液,加入500ml三颈圆底烧瓶中加热70℃进行混合溶解,时间约为1.3h。之后配制7g 1-氟-4-硝基苯浓度为75mmol,二甲基亚砜170ml、乙酸乙酯150ml继续添加到500ml三颈圆底烧瓶中,并在160℃氮气下反应30h。之后倒入700ml冷饱和盐水中,黄色沉淀物通过硅胶色谱法进行收集和纯化,并用二氯甲烷/己烷作为洗脱液,纯化后的产品为浅黄色针状晶体。
[0049]
s2、n、n-双(4-硝基苯基)-[1,1':4',1
”‑
三苯基]-3-胺的合成(n-tmbp):取用s1步骤的产物3g n-tmbr,配制为10-15mmol和使用2.5g 4-联苯硼酸浓度配制为12mmol添加到500ml三颈圆底烧瓶,加热44℃用索氏提取器进行冷凝回流。之后取用0.25g用50-70ml乙醇溶液溶解三苯基膦钯,并加入40ml k2co3水溶液与300ml四氢呋喃,最后加入在氮气下回流48h。去除水层后,通过旋转蒸发和蒸发收集黄色沉淀物硅胶,并用二氯甲烷层析纯化/己烷作为洗脱液。纯化产物为浅黄色针状晶体。
[0050]
s3、n
1-(1,1:4',1
”‑
三苯基]-3-基)-n
1-(4-氨基苯基)苯-1,4-二胺的合成(a-tmbp):取用步骤s2中得到的产物5g n-tmbp用乙酸乙酯配制成12mmol的溶液,并加入一匙0.07g 10%的钯/碳催化剂和200ml无水乙醇装入500ml三颈圆底烧瓶,然后,用滴液漏斗逐
滴添加4ml水合肼并在氮气下回流36小时。移除通过旋转蒸发乙醇,收集灰色沉淀物并然后用二氯甲烷在硅胶上层析纯化/己烷作为洗脱液。纯化产物为灰色晶体。
[0051]
s4、pi薄膜(tmbphf)的合成与制备:取用0.5g s3步骤的a-tmbp用乙酸乙酯溶解为2mmol的溶液,取用0.4g六氟二酐溶于乙醇使其配成2mmol溶液,并纯化将6ml二甲基甲酰胺添加到50ml烧瓶中,混合物是室温下在氩气下搅拌约5小时,形成粘性5-(2-(4-(1,1:4',1
”‑
三苯基)-3-基(4-氨基苯基)氨基)苯基)异吲哚-5-基)-1,1,1,3,3,3-六氟丙烷-2-基)异吲哚-1,3-二酮溶液(tmbphf)。随后在干净干燥的玻璃板上均匀涂上控制薄膜厚度,然后在真空中进行热亚胺化温度程序为150℃的烘箱,以生产pi薄膜。tmbphf膜冷却至室温后从玻璃基板上移除。
[0052]
对比例5除步骤s2中不加入4-联苯硼酸外,其余各步骤均与实施例3相同。
[0053]
对比例6除步骤s2中不加入4-联苯硼酸外,将4-联苯硼酸换为硼酸其余各步骤均与实施例3相同。
[0054]
表3
[0055]
检测项目对比例5对比例6实施例3co2选择性(%)10
±
0.0420
±
0.0180
±
0.02
[0056]
图5为本发明实施例3中a-tmbp有机化合物结构简式,图6为本发明实施例3中a-tmbp有机化合物核磁共振氢谱图。图9为本发明实施例3和对比例5、6所得到的co2选择性百分比柱状图。表3为本发明实施例3和对比例5、6co2选择性百分比平均数的统计表格。从图8和表2中可以看出实施例2中co2选择性以及捕集分离效果较强,高于对比例5、6。该结果表明,suzuki偶联反应引入的三联苯,可以在分子空间当中有效的进行旋转,有效地限制分子链的紧密堆积,在其周围产生更多的自由空间,更利于气体分子通过,从而改善聚酰亚胺膜材料的气体渗透性能。用多元共聚将刚性链锻和柔性链锻结合,刚性链段提供了主要的骨架结构,能够保持气体分离材料的热稳定性能,同时,柔性链段便于气体分子透过,保证气体的渗透性能。
[0057]
实施例4
[0058]
s1、3-溴-n,n-双(4-硝基苯)苯胺的合成(n-tmbr):取用6g的3-溴苯胺配制为30mmol的溶液,再取用10g氟化铯配制为60mmol的溶液,加入500ml三颈圆底烧瓶中加热70℃进行混合溶解,时间约为1.4h。之后配制7g 1-氟-4-硝基苯浓度为75mmol,二甲基亚砜170ml、乙酸乙酯180ml继续添加到500ml三颈圆底烧瓶中,并在160℃氮气下反应40h。之后倒入800ml冷饱和盐水中,黄色沉淀物通过硅胶色谱法进行收集和纯化,并用二氯甲烷/己烷作为洗脱液,纯化后的产品为浅黄色针状晶体。
[0059]
s2、n、n-双(4-硝基苯基)-[1,1':4',1
”‑
三苯基]-3-胺的合成(n-tmbp):取用s1步骤的产物6g n-tmbr,配制为12mmol和使用2-4g 4-联苯硼酸浓度配制为12mmol添加到500ml三颈圆底烧瓶,加热46℃用索氏提取器进行冷凝回流。之后取用0.4g用55ml乙醇溶液溶解三苯基膦钯,并加入37ml k2co3水溶液与250ml四氢呋喃,最后加入在氮气下回流70h。去除水层后,通过旋转蒸发和蒸发收集黄色沉淀物硅胶,并用二氯甲烷层析纯化/己烷作为洗脱液。纯化产物为浅黄色针状晶体。
[0060]
s3、n
1-(1,1:4',1
”‑
三苯基]-3-基)-n
1-(4-氨基苯基)苯-1,4-二胺的合成(a-tmbp):取用步骤s2中得到的产物5g n-tmbp用乙酸乙酯配制成11mmol的溶液,并加入一匙
0.07g 10%的钯/碳催化剂和300ml无水乙醇装入500ml三颈圆底烧瓶,然后,用滴液漏斗逐滴添加4.5ml水合肼并在氮气下回流36小时。移除通过旋转蒸发乙醇,收集灰色沉淀物并然后用二氯甲烷在硅胶上层析纯化/己烷作为洗脱液。纯化产物为灰色晶体。
[0061]
s4、pi薄膜(tmbphf)的合成与制备:取用0.4g s3步骤的a-tmbp用乙酸乙酯溶解为1.2mmol的溶液,取用0.5g六氟二酐溶于乙醇使其配成1.5mmol溶液,并纯化将6ml二甲基甲酰胺添加到50ml烧瓶中,混合物是室温下在氩气下搅拌约5小时,形成粘性5-(2-(4-(1,1:4',1
”‑
三苯基)-3-基(4-氨基苯基)氨基)苯基)异吲哚-5-基)-1,1,1,3,3,3-六氟丙烷-2-基)异吲哚-1,3-二酮溶液(tmbphf)。随后在干净干燥的玻璃板上均匀涂上控制薄膜厚度,然后在真空中进行热亚胺化温度程序为170℃的烘箱,以生产pi薄膜。tmbphf膜冷却至室温后从玻璃基板上移除。
[0062]
图10为本发明实施例4中tmbphf单体(pi薄膜单体)有机化合物结构简式,图11为本发明实施例4中tmbphf单体(pi薄膜单体)有机化合物核磁共振氢谱图。在大苯环侧基聚酰亚胺中引入的三氟甲基基团被互穿到聚合物主链上,增强了主链的刚性,同时三氟甲基的存在增加了侧基的体积,产生更多的自由空间,这将在保持高选择性的前提下,提高气体的渗透性;此外,三氟甲基基团还可提高气体的溶解性能,大体积苯环侧基通过增大空间位阻降低了堆积密度,利用-cf3基团和苯环侧基的协同作用增加了pi薄膜的自由体积,提高膜的气体渗透性能,实现了co2高效捕集和膜分离。
[0063]
最后应说明的是:以上所述的各实施例仅用于说明本发明技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分或全部技术特征进行等同替换;而这些修改或替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1