二聚异丁烯氢甲酰化催化剂组合物及其应用
戊烯的异构化-氢甲酰化反应速率,造成了异壬醛收率低、转化率低等问题,尤其是对于2,4,4-三甲基-2-戊烯比例较大的二聚异丁烯。
9.综上,现有的用于二聚异丁烯氢甲酰化反应的催化体系均存在反应条件苛刻、设备要求高、能耗高、反应活性低、催化剂稳定性差和流失量大等缺点。
技术实现要素:
10.本发明的一个目的在于提供二聚异丁烯氢甲酰化催化剂组合物,该催化剂组合物包括以一定比例混合的亚膦酰胺双膦配体和亚膦酰胺单膦配体,能够有效地增强催化剂活性物种铑的π电子接收能力和铑周围的空间效应,加速二聚异丁烯与铑金属的配位来提高2,4,4-三甲基-2-戊烯异构化-氢甲酰化的反应速率,进而提高了烯烃氢甲酰化反应的转化率和异壬醛的收率,同时,该催化剂组合物的稳定性高、易于反应后分离,能够在保持较高的收率、转化率的前提下循环利用,显著地降低了催化剂的投入成本。
11.本发明的此目的通过下述技术方案实现:
12.二聚异丁烯氢甲酰化催化剂组合物,包括第一膦配体和第二膦配体,所述第一膦配体具有式i所示的结构,所述第二膦配体具有式ii所示的结构:
[0013][0014]
式i、式ii中,r1、r2和r3各自独立地选自氢、烷基、环烷基、芳基、杂芳基、烷氧基、卤素、酰基、酯基、酰氧基、硝基、氰基。
[0015]
本技术方案中,催化剂组合物的膦配体包括以一定比例混合的第一膦配体和第二膦配体。其中,具有式i所示结构式的第一膦配体为亚膦酰胺双膦配体,其具有较强的螯合能力,能够更好地保护催化剂活性物种,即能催化中间烯烃的双键异构-氢甲酰化的串联反应的铑膦络物,增强铑催化剂的稳定性,进一步地有利于提高该催化体系的循环催化能力。但是,此类亚膦酰胺双膦配体在单独用于催化二聚异丁烯氢甲酰化反应时,二聚异丁烯氢甲酰化速率慢,这是由于在该铑/双膦催化体系下,金属铑周围的空间结构拥挤,导致较大位阻的2,4,4-三甲基-2-戊烯的双键(二聚异丁烯原料成分之一)异构生成2,4,4-三甲基-1-戊烯的速率慢。
[0016]
具有式ii所示结构式的第二膦配体为亚膦酰胺单膦配体,其有异构化功能和具有强π电子接收能力,能够有效地提高二聚异丁烯反应过程中的烯烃异构化-氢甲酰化的反应速率,但是单独用于二聚异丁烯氢甲酰化催化体系时,亚膦酰胺单膦配体的异壬醛选择性不高,且铑膦催化剂的稳定性差,催化体系难以循环利用,导致工业化应用成本高。
[0017]
在催化二聚异丁烯氢甲酰化反应过程中,二聚异丁烯中的2,4,4-三甲基-2-戊烯
先在铑膦复合催化剂作用下发生异构化生成2,4,4-三甲基-1-戊烯,再发生氢甲酰化反应生成异壬醛。
[0018][0019]
发明人在研究过程中发现,当采用特定组合的亚膦酰胺单膦配体和亚膦酰胺双膦配体组合应用于二聚异丁烯氢甲酰化反应中时,采用铑催化剂前体与具有强π电子接收能力的p-n结构模型的第一配体和第二配体形成特定组合催化剂的策略,增强了催化剂活性物种铑电子接收能力和铑周围的空间效应,加速了二聚异丁烯与铑金属的配位,进而提高了2,4,4-三甲基-2-戊烯异构化的反应速率,提高了异壬醛的收率,且在循环催化过程中,该催化剂组合能够加强催化剂活性物种铑的稳定性和催化活性,在多次循环催化过程中均能保持较高的转化率和异壬醛选择性。
[0020]
作为本发明中第一膦配体的优选实施方式,所述第一膦配体具有以下任一种结构式:
[0021][0022]
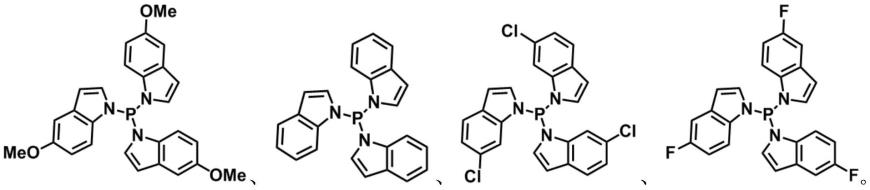
作为本发明中第二膦配体的优选实施方式,所述第二膦配体具有以下任一种结构式:
[0023][0024]
进一步地,所述第一膦配体和第二膦配体的摩尔比为1:10~1:80。通过实验发现,当第一膦配体和第二膦配体的摩尔比在1:10~1:80时,包含该催化剂组合物的催化剂溶液不仅能够确保80%以上的转化率,异壬醛的选择性高达98%,而且可以在多次循环过程中均保持较高的转化率和异壬醛的选择性。当第一膦配体过少或第二膦配体过多时,二聚异丁烯氢甲酰化反应的转化率将会降低,且保持高转化率、高选择性的循环催化次数也将减少。优选地,第一膦配体和第二膦配体的摩尔比为1:20~1:80,进一步优选地,第一膦配体和第二膦配体的摩尔比为1:20~1:40。
[0025]
进一步地,还包括铑催化剂前体,所述第一配体与所述铑催化剂前体的摩尔比为0.5~10,所述第二配体与所述铑催化剂前体的摩尔比为20~80。催化剂组合物中还包括有
铑催化剂前体,其中,第一配体与所述铑催化剂前体的摩尔比为0.5~10,优选为0.5~4,进一步优选为1~2;第二配体与所述铑催化剂前体的摩尔比为20~80,优选为20~40,进一步优选为30~40。
[0026]
进一步地,铑催化剂前体为rh(acac)(co)2、[rh(co)2]2cl2、rh(cod)2bf4、rh(nbd)2bf4、rh(cod)(acac)、hrh(co)(tpp)3、rhcl3、[rh(cod)cl]2、rh(c2h4)(acac)、[rh(c2h4)cl]2、rh(oac)3、rh2(oac)4、rh(no3)3、[rh(c7h
15
coo)2]2中的至少一种,其中,acac为乙酰丙酮基,cod为1,5-环辛二烯,nbd为降冰片二烯,tpp为三苯基膦,oac为醋酸根离子。优选地,所述铑催化剂前体为rh(acac)(co)2。
[0027]
本发明的另一个目的在于基于前述任一种二聚异丁烯氢甲酰化催化剂组合物,提供该催化剂组合物在二聚异丁烯氢甲酰化反应中的应用,不仅能够提高2,4,4-三甲基-2-戊烯异构化-氢甲酰化的反应速率,保持较高的催化剂使用寿命,在多次循环中保持较高的收率、转化率,而且反应条件温和,满足工业规模化生产。
[0028]
本发明的此目的通过下述技术方案实现:
[0029]
基于前述任一种催化剂组合物在二聚异丁烯氢甲酰化反应中的应用,所述应用包括以下步骤:
[0030]
将所述催化剂组合物、二聚异丁烯、溶剂混合均匀后形成反应液,所述反应液在高温高压下与一氧化碳和氢气构成的合成气反应;
[0031]
反应完成后,通过减压蒸馏方式分离产物与催化剂溶液,所述催化剂溶液能够重复用于催化新加入的二聚异丁烯。
[0032]
本技术方案中,向高压反应釜中加入铑催化剂前体、第一膦配体、第二膦配体构成的催化剂组合物,2,4,4-三甲基-1-戊烯与2,4,4-三甲基-2-戊烯以任意比例混合的二聚异丁烯,以及溶剂。闭釜后,利用一氧化碳与氢气构成的合成器置换反应釜内空气数次后,充入适量的合成气以使反应釜内保持一定的压力,在高温高压下反应一段时间后,蒸馏分离得到产物醛,此时釜底的催化剂溶液仍然具有较高的活性和稳定性,能够用于循环催化反应。循环催化反应的步骤与前述相同,每次循环反应中无需再加入催化剂组合物,利用蒸馏分离后釜内剩余的催化剂组合物催化新加入的二聚异丁烯氢甲酰化反应即可。
[0033]
在一个或多个实施例中,所述溶剂为3,5,5-三甲基己醛,3,5,5-三甲基己醇,二甲苯,二乙二醇二丁醚,四乙二醇二甲醚,1-辛醇,1-壬醇,1-癸醇,二乙二醇,三乙二醇,四乙二醇中的至少一种。
[0034]
在一个或多个实施例中,所述催化反应形式为间歇式或连续式反应,反应时间为3小时~10小时,优选为6小时~8小时。
[0035]
在一个或多个实施例中,反应结束后采取分步分离的方法,先在45℃~50℃/-0.09mpa条件下蒸出未反应的二聚异丁烯,然后,在80℃~90℃/-0.09mpa~-0.095mpa条件下蒸馏出产物异壬醛。分离出的二聚异丁烯可作为原料继续投入反应,与产品分离后的催化剂溶液加入原料二聚异丁烯后继续重复使用。
[0036]
在一个或多个实施例中,合成气的组成为h2与co体积比为1:0.9~1.5:1,优选为1:1~1.2:1。
[0037]
进一步地,所述催化剂组合物中的铑催化剂前体与所述二聚异丁烯的摩尔比为1:500~1:10000。优选地,催化剂溶液中铑浓度为50ppm~600ppm,优选为200ppm~400ppm。
[0038]
进一步地,所述反应温度为70~100℃,所述反应压力为2~5mpa。应用该催化剂组合物的二聚异丁烯氢甲酰化反应的反应温度可以介于60~120℃之间,优选为70~100℃,进一步优选为80~90℃。恒压反应的压力可以介于1~8mpa,优选为2~5mpa,进一步优选为3~5mpa。
[0039]
进一步地,所述催化剂溶液能够重复循环催化二聚异丁烯至少5次,且每次循环催化反应的烯烃转化率大于95%,异壬醛选择性大于97%。
[0040]
本发明与现有技术相比,具有如下的优点和有益效果:
[0041]
1、本发明提供的催化剂组合物包括特定组合的亚膦酰胺单膦配体和亚膦酰胺双膦配体,当其用于二聚异丁烯氢甲酰化反应中时,具有强π电子接收能力的p-n结构模型的第一配体和第二配体增强了催化剂活性物种铑电子接收能力和铑周围的空间效应,加速了二聚异丁烯与铑金属的配位,进而提高了2,4,4-三甲基-2-戊烯异构化的反应速率,提高了异壬醛的收率,且在循环催化过程中,该催化剂组合能够加强催化剂活性物种铑的稳定性和催化活性,在多次循环催化过程中均能保持较高的转化率和异壬醛选择性,解决了三取代烯烃氢甲酰化反应活性差的难题,同时提供了解决中间烯烃异构化-氢甲酰化反应速率低的新催化方法,二聚异丁烯氢甲酰化反应转化率大于95%,异壬醛选择性大于97%;
[0042]
2、当第一膦配体过少或第二膦配体过多时,二聚异丁烯氢甲酰化反应的转化率将会降低,且保持高转化率、高选择性的催化剂循环使用次数也将减少,因此,本发明将第一膦配体和第二膦配体的摩尔比在1:10~1:80时,包含该催化剂组合物的催化剂溶液不仅能够确保80%以上的烯烃转化率,异壬醛的选择性高达98%,而且可以在多次循环过程中均保持较高的转化率和异壬醛选择性;
[0043]
3、本发明提供的催化剂组合物在二聚异丁烯氢甲酰化反应中的应用,不仅能够提高2,4,4-三甲基-2-戊烯异构化-氢甲酰化的反应速率,保持较高的催化剂使用寿命,在多次循环中保持较高的收率、转化率,而且反应条件温和,适用于工业规模化生产。
具体实施方式
[0044]
为使本发明的目的、技术方案和优点更加清楚明白,下面结合实施例,对本发明作进一步的详细说明,本发明的示意性实施方式及其说明仅用于解释本发明,并不作为对本发明的限定。
[0045]
本发明所有原料,对其来源没有特别限制,在市场上购买的或按照本领域技术人员熟知的常规方法即可制备。
[0046]
本发明所有原料,对其纯度没有特别限制,本发明优选采用分析纯或二聚异丁烯氢甲酰化领域常规的纯度要求。
[0047]
本发明所有原料,其牌号和简称均属于本领域常规牌号和简称,每个牌号和简称在其相关用途的领域内均是清楚明确的,本领域技术人员根据牌号、简称以及相应的用途,能够从市售中购买得到或者通过常规方法制备得到。
[0048]
本发明对所述取代基的表达方式没有特别限制,均采用本领域技术人员熟知的表达方式,本领域技术人员基于常识,可根据其表达方式正确理解其含义。
[0049]
本发明所使用的“第一”、“第二”等(例如第一膦配体、第二膦配体等)只是为了描述清楚起见而对相应组分/材料/化合物进行区别,不旨在限制任何次序或者强调重要性
(34.4mmol)和10.5ml(2.2eq.,75.7mmol)无水无氧的et3n,并将其在冰水浴中冷却至0℃。向100ml干燥后的恒压滴液漏斗中依次加入8.06g吲哚(2.0eq.,68.8mmol)和40ml无水无氧的thf。在0℃且剧烈搅拌的条件下,将恒压滴液漏斗中的混合溶液在30min内缓慢滴加到三颈瓶中,一经滴加立即可见白色et3n
·
hcl固体产生于瓶底。当滴加完成后,采取程序式升温,先维持0℃的条件下强力搅拌0.5h,然后移除冰浴,使反应液自然升温至环境温度。反应液不经任何处理直接用于下一步反应。
[0062]
向上述100ml恒压滴液漏斗中分别加入3.2g 2,2
′
,-二羟基-1,1
′
,-联萘和30ml无水无氧的thf,再向三颈瓶中补加4.5ml无水无氧的et3n(2.2eq.,32.2mmol),将溶液冷却至0℃,30min内将恒压滴液漏斗中的2,2
′
,-二羟基-1,1
′
,-联萘溶液缓慢滴加至上述含二吲哚氯膦的溶液中。滴加完毕后,保持0℃继续强力搅拌0.5h,随后移走冰浴使其缓慢自然升至环境温度,然后缓慢加热至40℃。维持40℃的条件下继续强力搅拌12h。反应完成后,停止加热反应液,缓慢降至环境温度。过滤得到淡黄色滤液,缓慢滴加30ml无水乙醇重结晶得到9.2g粗产物。经过第二次重结晶得6.6g晶状固体,即为第一膦配体b:
[0063][0064]
核磁表征数据:1h nmr(400mhz,cdcl3)δ7.81(d,j=8.2hz,2h),7.71(d,j=8.9hz,2h),7.54(d,j=7.8hz,2h),7.47(d,j=7.8hz,2h),7.40(ddd,j=8.1,6.3,1.6hz,2h),7.29-7.21(m,6h),7.19(d,j=8.2hz,2h),7.10(qd,j=7.9,0.7hz,6h),7.03-6.92(m,4h),6.80-6.70(m,4h),6.39(d,j=3.3hz,2h),6.33(d,j=3.4hz,2h).
[0065]
31
p nmr(162mhz,cdcl3)δ104.71.
[0066]
二、第二膦配体的合成
[0067][0068]
其中,为吡咯结构衍生物,r选自芳环、芳杂环、芳基、烷基、环烷基、环杂烷基、氢等,包括吡咯、吲哚、咔唑及其衍生物等。
[0069]
【实施例3】
[0070]
第二膦配体1:三(5-甲氧基吲哚基)膦配体的合成:
[0071]
在0℃,n2保护下,向1000ml三口圆底烧瓶中依次加入600ml经分子筛干燥处理的四氢呋喃、120.9g 5-甲氧基吲哚(1031.5mmol)和170ml(1237.8mmol)经分子筛干燥处理的三乙胺溶液。在剧烈搅拌条件下用注射器将30ml三氯化磷(343.8mmol)分批多次加入到上述含有5-甲氧基吲哚和三乙胺的溶液中,一经滴加立即产生大量白色三乙胺盐酸盐固体,形成悬浊液。待三氯化磷完全加入到反应体系,继续保持0℃半小时后移除冰浴,使反应液自然升温至环境温度。而后强力搅拌过夜,最终得到略带淡黄色的反应液。过滤、洗涤、去除溶剂,加入不良溶剂进行重结晶得到略带淡黄色的白色固体,经过滤干燥后最终得到129.1g白色固体,收率为80%,即第二膦配体1:
[0072][0073]
核磁表征数据:1h nmr(400mhz,cdcl3)δ7.38(d,j=8.9hz,3h),7.09(d,j=1.1hz,3h),6.94(t,j=3.2hz,3h),6.84(dd,j=9.0,2.5hz,3h),6.62(d,j=3.4hz,3h),3.84(s,9h).
[0074]
31
p nmr(162mhz,cdcl3)δ68.35.
[0075]
【实施例4】
[0076]
第二膦配体2:三吲哚基膦配体的合成:
[0077]
在0℃,n2保护下,向1000ml三口圆底烧瓶中依次加入600ml经分子筛干燥处理的四氢呋喃、120.6g吲哚(1031.5mmol)和170ml(1.2*1031.5mmol)经分子筛干燥处理的三乙胺(缚酸剂,一般用有机碱)形成溶液。在剧烈搅拌条件下用注射器将30ml三氯化磷(343.8mmol)分多次加入到上述含有吲哚和三乙胺的溶液中,一经滴加立即产生大量白色三乙胺盐酸盐固体,形成悬浊液。待三氯化磷完全加入到反应体系,继续保持0℃半小时后移除冰浴,待反应液自然升温至环境温度后。而后强力搅拌过夜,最终得到略带淡黄色的反应液。过滤、洗涤、去除溶剂,加入不良溶剂进行重结晶得到略带淡黄色的白色固体,经过滤干燥后最终得到97.8g白色固体,75%收率,即第二膦配体2:
[0078][0079]
核磁表征数据:
[0080]1h nmr(400mhz,cdcl3)δ7.79-7.62(m,6h),7.37-7.26(m,6h),7.10-7.01(m,3h),6.78(d,j=3.4hz,3h).
[0081]
31
p nmr(162mhz,cdcl3)δ66.82.
[0082]
【实施例5】
[0083]
第二膦配体3:三(6-氯吲哚基)膦配体得合成:
[0084]
在0,℃n2保护下,向1000ml三口圆底烧瓶中依次加入600ml经分子筛干燥处理的四氢呋喃、156.4g吲哚(1031.5mmol)和170ml(1.2*1031.5mmol)经分子筛干燥处理的三乙胺(缚酸剂,一般用有机碱)形成溶液。在剧烈搅拌条件下用注射器将30ml三氯化磷(343.8mmol)分多次加入到上述含有吲哚和三乙胺的溶液中,一经滴加立即产生大量白色三乙胺盐酸盐固体,形成悬浊液。待三氯化磷完全加入到反应体系,继续保持0℃半小时后移除冰浴,待反应液自然升温至环境温度后。而后强力搅拌过夜,最终得到略带淡黄色的反应液。过滤、洗涤、去除溶剂,加入不良溶剂进行重结晶得到略带淡黄色的白色固体,经过滤干燥后最终得到三(6-氯吲哚基)膦配体117.8g,收率为71%,即第二膦配体3:
[0085][0086]
核磁表征数据:1h nmr(400mhz,cdcl3)δ7.61
–
7.51(m,6h),7.22(dd,j=8.5,1.7hz,3h),6.92
–
6.86(m,3h),6.70(d,j=3.4hz,3h).
[0087]
31
p nmr(162mhz,cdcl3)δ66.25.
[0088]
【实施例6】
[0089]
第二膦配体4:三(5-氟吲哚基)膦配体
[0090]
在0,℃n2保护下,向1000ml三口圆底烧瓶中依次加入600ml经分子筛干燥处理的四氢呋喃、139.2g吲哚(1031.5mmol)和170ml(1.2*1031.5mmol)经分子筛干燥处理的三乙胺(缚酸剂,一般用有机碱)形成溶液。在剧烈搅拌条件下用注射器将30ml三氯化磷(343.8mmol)分多次加入到上述含有吲哚和三乙胺的溶液中,一经滴加立即产生大量白色三乙胺盐酸盐固体,形成悬浊液。待三氯化磷完全加入到反应体系,继续保持0℃半小时后移除冰浴,待反应液自然升温至环境温度后。而后强力搅拌过夜,最终得到略带淡黄色的反应液。过滤、洗涤、去除溶剂,加入不良溶剂进行重结晶得到略带淡黄色的白色固体,经过滤干燥后最终得到三(5-氟吲哚基)膦配体112.0g,收率为78%,即第二膦配体4:
[0091][0092]
核磁表征数据:1h nmr(400mhz,cdcl3)δ8.08(d,j=7.4hz,6h),7.29
–
7.24(m,6h),7.15(ddd,j=12.4,9.5,4.6hz,12h).
[0093]
31
p nmr(162mhz,cdcl3)δ67.63.
[0094]
三、二聚异丁烯氢甲酰化反应
[0095][0096]
【实施例7~17】
[0097]
向50ml高压反应釜中分别加入rh(acac)(co)2与第一膦配体和第二膦配体形成的组合催化剂,铑浓度=200~300ppm,第一膦配体与rh的摩尔比为0.5~10,第二膦配体与rh的摩尔比为20~80,40ml二聚异丁烯(s/c=8200),闭釜。合成气置换反应釜内空气三次后,充入适量合成气,加热至反应温度后,保持一定压力恒压反应,反应时间为6h,实验结果如表1所示:
[0098]
表1:
[0099][0100]
由表1可知,本发明提供的催化剂组合有效地提高了二聚异丁烯氢甲酰化的反应速率和醛的选择性,特别是提供了最佳的第一膦配体与第二膦配体的摩尔比值时,与铑前体形成的催化剂的催化活性最高,二聚异丁烯转化率达到96%以上。进一步地,在第一膦配体与第二膦配体的摩尔比值为2:40的优选条件下,不同的第一膦配体(a、b)与第二膦配体(1、2、3、4)进行交叉组合后,均能在二聚异丁烯氢甲酰化反应中保持较高的催化活性,其中二聚异丁烯转化率达到96%,异壬醛的选择性达到98%。
[0101]
【实施例18】
[0102]
采用第一膦配体a、第二膦配体1、rh(acac)(co)2构成的催化剂组合物,用于二聚异丁烯的循环催化反应。首次催化反应结束后,分离得到的产物醛,釜底催化剂溶液继续循环利用5次,实验结果如表2所示:
[0103]
表2:
[0104][0105][0106]
由表2可知,采用的第一膦配体a和第二膦配体1组合与铑催化剂前体催化二异丁烯氢甲酰化反应,循环使用5次后,转化率和选择性均无明显变化,每次循环催化反应的烯烃转化率大于95%,异壬醛选择性大于97%,这是由于第一膦配体a和第二膦配体1的组合共同调控了催化剂活性物种铑接收电子能力和铑周围的空间效应,能够有效稳定铑催化剂活性物种。
[0107]
通过对比例1可知,当单独采用第一膦配体a与铑前体催化二聚异丁烯氢甲酰化反应时,循环使用5次过程中,烯烃转化率基本稳定在75%左右,明显较以第一膦配体a和第二膦配体1的组合作为催化剂组合物的催化效果低;同时,通过对比例2可知,采用第二膦配体1与铑前体催化二聚异丁烯氢甲酰化反应,在循环过程中,催化剂活性急剧下降,该催化剂循环使用5次后,基本没有催化活性,推测第二膦配体1与铑的螯合能力差,不能有效地稳定铑膦催化活性物种,导致佬催化活性物种转变为非活性物种的铑膦化合物。
[0108]
【实施例19】
[0109]
催化反应结束后,未反应完烯烃原料进行蒸馏分离,分离温度为40℃~45℃,分离真空度为-0.09mpa~-0.095mpa,得到的烯烃再次投入反应釜中进行反应,反应条件:[rh]=300ppm,第一膦配体a和第二膦配体1,第一膦配体a/rh=2,第二膦配体1/rh=40,40ml二聚异丁烯(81%2,4,4-三甲基-1-戊烯和19%2,4,4-三甲基-2-戊烯混合物,s/c=8200),加热至90℃,保持合成气5mpa恒压,反应时间为6h。实验结果如表3所示:
[0110]
表3
[0111][0112]
由表3可知,本发明采用的第一膦配体a和第二膦配体1组合与铑催化剂前体催化二异丁烯氢甲酰化反应过程中,2,4,4-三甲基-2-戊烯在铑膦催化剂作用下发生异构化形成2,4,4-三甲基-1-戊烯,在发生氢甲酰化生成异壬醛。
[0113]
以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1