一种GO基恶唑型混合基质膜及其制备方法与流程
一种go基恶唑型混合基质膜及其制备方法
技术领域
1.本发明属于高分子材料领域,尤其是涉及一种go基恶唑型混合基质膜及其制备方法。
背景技术:
2.随着工业经济的快速发展,全世界的化石能源消耗与日俱增。大气中co2的浓度过高,引起了极端气候频发、海平面不断上升、土地沙漠化、物种多样化减少等危害。因此对于co2的分离与捕集是一件刻不容缓的任务。
3.对于co2的分离技术主要有吸收、吸附、低温精馏、膜分离法,其中膜分离法因其能耗低、占地面积小、环境友好等优势成为未来分离技术的主导。在众多膜材料中,混合基质膜能够综合利用填料及基膜的两种优势,相比于纯膜,其对于co2的渗透通量和co2的选择性,得到极大提升。
4.聚酰亚胺(pi)是一种主链含有亚酰胺环的高分子材料,被誉为“21世纪最有希望的工程塑料之一”,位于塑料材料的金字塔顶端。具有优异的热稳定性、耐化学、良好的机械性能及疏水性能。以此作为基膜能够应用于各种苛刻的条件下,基膜本身具有良好的co2/ch4选择性。石墨烯(go)是一种二维材料,在片层中存在一定的空隙。以go作为填料,其空隙能够为气体的渗透提供孔道,增强其co2的渗透通量。
5.目前,以聚酰亚胺作为基膜,综合性能较为优异,但是co2的渗透通量偏低。引入填料go进行掺杂,能够极大程度上解决这一问题。但是由于go是一种无机材料,利用纯go进行掺杂制备混合基质膜时,会发生界面缺陷,产生过多的非选择性孔隙,降低co2的选择性。
技术实现要素:
6.有鉴于此,本发明旨在提出一种go基恶唑型混合基质膜及其制备方法,以解决界面缺陷问题,改善分离性能。
7.为达到上述目的,本发明的技术方案是这样实现的:
8.一种go基恶唑型混合基质膜的制备方法,包括以下步骤:
9.s1、制备2-(4-氨基苯基)-5-氨基苯并恶唑;
10.s2、制备恶唑型聚酰亚胺;
11.s3、制备功能化石墨烯;
12.s4、将恶唑型聚酰亚胺、功能化石墨烯与dmac混合均匀得到铸膜液,将铸膜液涂抹在模板上,干燥得到预制膜,将预制膜高温处理,得到所需混合基质膜。
13.本发明的发明构思在于,首先合成2-(4-氨基苯基)-5-氨基苯并恶唑(apboa),与六氟二酐(6fad)合成恶唑型聚酰亚胺,恶唑环与co2具有较强的亲和力,因此6fda-apboa型聚酰亚胺具有很高的co2/ch4选择性,go经过离子液体功能化后,解决了界面缺陷问题,由此制备的混合基质膜,具有非常优异的co2分离性能。
14.进一步地,步骤s1中制备2-(4-氨基苯基)-5-氨基苯并恶唑的方法包括以下步骤:
15.s11、将对硝基苯甲酰氯溶液降温后,加入2-氨基-5-硝基苯酚与缚酸剂的混合溶液进行反应,得到反应液;
16.s12、过滤反应液,将得到的固体干燥后溶于第一溶剂中,加入第一催化剂,加热至反应完全,蒸干溶剂,得到2-(4-硝基苯基)-5-硝基苯并恶唑;
17.s13、将2-(4-硝基苯基)-5-硝基苯并恶唑溶于乙醇后,加入钯碳搅拌均匀,通入氢气至反应完全,去除钯碳及乙醇,得到2-(4-氨基苯基)-5-氨基苯并恶唑。
18.进一步地,所述第一催化剂为三氟甲磺酸、浓硫酸、多聚磷酸中的一种或多种。
19.进一步地,钯碳与2-(4-硝基苯基)-5-硝基苯并恶唑的质量比为0.01-0.05:1;优选地,所述钯碳为5wt%。
20.进一步地,步骤s2中制备恶唑型聚酰亚胺的方法包括以下步骤:
21.s21、将六氟二酐、2-(4-氨基苯基)-5-氨基苯并恶唑与dmac混合反应后,加入脱水剂及第二催化剂,继续反应至完全,得到反应液;
22.s22、将反应液加入不良溶剂中,将析出的固体过滤、洗涤、干燥,得到所需恶唑型聚酰亚胺。
23.进一步地,所述不良溶剂为去离子水、甲醇、乙醇、异丙醇中的一种;优选为甲醇或乙醇;更优选为乙醇。
24.进一步地,步骤s3中制备功能化石墨烯的方法包括以下步骤:
25.将离子液体与第二溶剂混合均匀,加入石墨烯,搅拌均匀后蒸干溶剂,得到功能化石墨烯。
26.进一步地,所述离子液体为1,3-二甲基咪唑四氟硼酸盐、1-乙基-3-甲基咪唑硫酸甲酯盐中的一种或两种的溶液。
27.进一步地,步骤s4中铸膜液的固含量为5wt%-10wt%;优选地,预制膜高温处理温度为180-220℃。
28.根据如上任一所述的制备方法制得的go基恶唑型混合基质膜。
29.相对于现有技术,本发明所述的go基恶唑型混合基质膜及其制备方法具有以下优势:
30.本发明所述的go基恶唑型混合基质膜具有较高的co2/ch4的选择性,以功能化的go作为填料制备的混合基质膜进一步提升了co2的分离性能,制备方法简单,环境友好,co2分离性能优异,为工业化提供一定的借鉴性。
附图说明
31.构成本发明的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
32.图1为本发明实施例所述的2-(4-氨基苯基)-5-氨基苯并恶唑的合成路线示意图;
33.图2为本发明实施例1制得的2-(4-硝基苯基)-5-硝基的hplc示意图;
34.图3为本发明实施例1制得的2-(4-硝基苯基)-5-氨基的hplc示意图;
35.图4为本发明测试例使用的分离膜的气体分离测试仪器示意图;
36.图5为本发明测试例的测试结果示意图。
具体实施方式
37.需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
38.下面将参考附图并结合实施例来详细说明本发明。
39.实施例1
40.2-(4-硝基苯基)-5-硝基苯并恶唑的合成
41.称取19.48g对硝基苯甲酰氯溶于50ml乙腈中,降温至0℃;再将15.41g的2-氨基-4-硝基苯酚和6.96g环氧丙烷溶于40ml乙腈中,然后将其滴加到上述溶液中;滴加完毕后,室温反应6h。产品析出,将其过滤后,80℃真空干燥5h;将烘干后固体加入到200mlγ-丁内酯中,加入9.23g浓h2so4,搅拌均匀后,升温至80℃反应12h,将溶剂蒸干后,利用500ml去离子水洗涤后,110℃真空干燥18h,得到27.49g目标硝基物,摩尔收率96.4%,纯度为99.92%。
42.2-(4-氨基苯基)-5-氨基苯并恶唑的合成
43.将20g的2-(4-硝基苯基)-5-硝基、0.3g钯碳和150g乙醇加入到500ml高压釜中,搅拌均匀后,将温度升至60℃。通入1mpa h2,保温反应12h。反应结束后,热过滤除去钯碳得到滤液,减压蒸馏回收100g乙醇,降温冷却至0℃,有大量白色晶体析出,过滤后得到固体,在60℃下真空干燥8h,得到15.4g产品,摩尔收率为97.51%,纯度为99.95%。
44.实施例2
45.2-(4-硝基苯基)-5-硝基苯并恶唑的合成
46.称取19.87g对硝基苯甲酰氯溶于50ml乙腈中,降温至0℃。再将15.41g的2-氨基-4-硝基苯酚和9.49g吡啶溶于40ml乙腈中。然后将其滴加到上述溶液中。滴加完毕后,室温反应7h。产品析出,将其过滤后,利用200ml去离子水洗涤,100℃真空干燥5h;将烘干后固体加入到200ml二氧六环中,加入21.23g多聚磷酸,搅拌均匀后,升温至80℃反应12h,将溶剂蒸干后,利用500ml去离子水洗后,110℃真空干燥18h,得到27.03g目标硝基物,摩尔收率94.8%,纯度为99.73%。
47.2-(4-氨基苯基)-5-氨基苯并恶唑的合成
48.将20g的2-(4-硝基苯基)-5-硝基、0.25g钯碳和150g乙醇加入到500ml高压釜中,搅拌均匀后,将温度升至60℃。通入0.8mpa h2,保温反应12h。反应结束后,热过滤除去钯碳得到滤液,减压蒸馏回收100ml乙醇,降温冷却至0℃,有大量白色晶体析出,过滤后得到固体,在60℃下真空干燥8h,得到15.28g产品,摩尔收率为96.74%,纯度为99.85%。
49.实施例3
50.将10g六氟二酐加入到30ml二甲基乙酰胺中搅拌溶解后,降温至0℃,再将5.07g apboa加到反应页中,保温反应8h,再将9.19g乙酸酐和2.28g三乙胺加到反应体系中升温至25℃反应6h。将其倒入500ml乙醇中,有大量丝状固体析出,将其过滤,再次利用500ml乙醇洗涤8h后,真空130℃真空干燥24h,得到13.53g恶唑型聚酰亚胺6fda-apboa。
51.实施例4
52.将9.97g六氟二酐加入到30ml二甲基乙酰胺中搅拌溶解后,降温至0℃,再将5.07g apboa加到反应液中,保温反应8h,再将2.93g丙酸酐和1.78g吡啶加到反应体系中升温至25℃反应6h。将其倒入500ml异丙醇中,有大量丝状固体析出,将其过滤,再次利用500ml异丙
醇洗涤8h后,真空130℃真空干燥24h,得到12.98g恶唑型聚酰亚胺6fda-apboa。
53.实施例5
54.将200mg的离子液体1,3-二甲基咪唑四氟硼酸盐溶于10ml四氢呋喃中,向其中加入100mg的go搅拌3h,超声3h后。将其倒入烧杯中,在50℃下缓慢搅拌,持续1h缓慢蒸干,将固体60℃真空干燥8h,取出后得到260mg功能化石墨烯il-go-1。
55.实施例6
56.将200mg的离子液体,1-乙基-3-甲基咪唑硫酸甲酯盐溶于10ml二氯甲烷中,向其中加入100mg的go搅拌3h,超声3h后。将其倒入烧杯中,在50℃下缓慢搅拌,持续1h缓慢蒸干,将固体60℃真空干燥8h,取出后得到280mg功能化石墨烯il-go-2。
57.实施例7
58.将0.5g的恶唑型聚酰亚胺6fda-apboa溶于7ml dmac中,搅拌2h,超声3h脱出气泡后,涂抹到四氟乙烯板上,在加热平台上50℃放置8h,固定成为预制膜。剥离后在真空烘箱中200℃真空干燥24h,得到分离膜,6fda-apboa。
59.将0.5g的恶唑型聚酰亚胺6fda-apboa溶于7ml dmac中,再将2mg il-go-1研磨后掺杂到铸膜液中,搅拌2h,超声3h脱出气泡后,涂抹到四氟乙烯板上,在加热平台上50℃放置8h,固定成为预制膜。剥离后在真空烘箱中200℃真空干燥24h,得到分离膜,il-go-1/6fda-apboa。
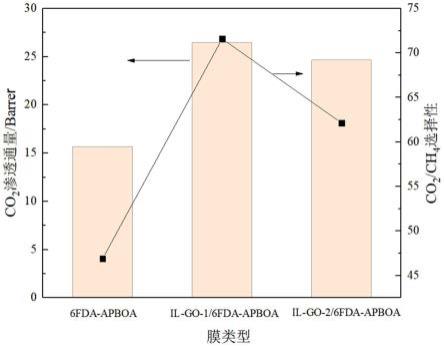
60.实施例8
61.将0.5g的恶唑型聚酰亚胺6fda-apboa溶于7ml dmac中,再将2mg il-go-2研磨后掺杂到铸膜液中,搅拌2h,超声3h脱出气泡后,涂抹到四氟乙烯板上,在加热平台上50℃放置8h,固定成为预制膜。剥离后在真空烘箱中200℃真空干燥24h,得到分离膜,il-go-2/6fda-apboa。
62.测试例
63.对实施例7-8中制得的分离膜进行co2/ch4气体分离性能实验,气体分离性能测试设备连接结构如图4所示,其中f1-f10均为阀门,具体实验步骤如下:
64.(1)将实例7-8中制备得到的分离膜,分别装入膜池后,先开真空泵、f3、f6、f7,再开f4、f5,接着缓慢开启f8进行上下膜池同时抽气,此时f1、f2处于关闭状态;当真空度达3.0
×
10-2
torror以下时即可测试;
65.(2)首先依次关上f4、f8,然后开f1充入一定压力的测试气体,反复开f2置换气体三次,调整好进样压力后,关闭f3(根据测试气体需要)、f6、f7、真空泵,开f4点击“start”开始数据采集,此时f1、f2仍然处于关闭状态;
66.(3)测试结束后开真空泵、f3、f6、f7,关减压阀,开f1、f2排空进样管路气体后,关f1、f2,再缓慢开启f8同时抽气。
67.通过测试上膜池的气体压力随着时间变化的曲线,即可计算出气体的渗透系数p。如图5所示,按照本发明提供的技术方案,得到的恶唑型聚酰亚胺(6fda-apboa)作为基膜,本身具有优异的co2分离性能,利用功能化go作为填料制备的混合基质膜具有较高co2渗透通量和高的co2/ch4选择性。
68.6fda-apboa作为基膜,co2的渗透通量为15.67barrer,co2/ch4的选择性为46.89。相比于基膜,il-go-1/6fda-apboa的渗透通量提升了68.94%,co2/ch4的选择性提升了
52.67%,il-go-2/6fda-apboa的渗透通量提升了57.38%,co2/ch4的选择性提升了32.54%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1