一种双载体上负载钯镍纳米催化剂的制备方法及应用
1.本发明涉及催化剂制备以及环境和能源的可持续发展领域,特别是一种双载体上负载钯镍纳米催化剂的制备方法及应用。
背景技术:
2.可持续发展是全球关注的主题,人与自然的和谐是发展的基础。氢能因其燃烧热值高、清洁无污染和来源广泛等优点,被认为是一种非常有前景的能量载体。但是,氢能的安全存放和运输仍然是面临的严峻挑战之一。近年来,化学储氢材料引起了广泛的关注。甲酸(hcooh,fa)由于具有高能量密度(4.4wt%),无毒和稳定性良好等优点被认为是一种安全方便的液相储氢材料。在使用合适的催化剂和在合适的反应条件,甲酸可以通过优选的制氢途径(hcooh
→
co2+h2,
△g298k
=-48.8kj mol-1
)释放存储的氢。然而,制氢过程中发生副反应(hcooh
→
co+h2o,
△g298k
=-28.5kj mol-1
)会产生一氧化碳,对催化剂有毒。
3.一般来说,在均相催化体系和多相催化体系中均可以实现液相储氢材料制氢。然而,均相催化体系存在快速失活、分离和再循环困难等缺点,阻碍了它们的实际应用。近年来,钯(pd)基多相催化剂在甲酸分解领域取得了很大进展。pd是对甲酸制氢反应最活跃的金属之一。然而,纯pd催化剂吸附co容易失活,并且高成本阻碍了其大规模应用。为了解决这些问题,研究者们考虑掺入非贵金属(如fe、co、ni等)形成多金属pd基催化剂。通常由于活性位点的增加,尺寸小的金属纳米颗粒可以提高制氢效率。然而,金属纳米颗粒一般具有较高的表面自由能,因此,在催化过程中金属纳米颗粒容易团聚而导致长期稳定性和回收性差。将金属纳米颗粒固定在具有高比表面积的载体上,可以通过强金属-载体相互作用进一步提高催化活性。例如专利cn113426469a,一种用于甲酸制氢的双载体支撑镍钯纳米催化剂的制备方法及应用,将mofs衍生的多孔碳和还原氧化石墨烯复合,可以很大程度上提高甲酸催化性能,但是其循环性仅有4次。再如cn113042086a,一种氨基功能化碳纳米管负载niaupd纳米催化剂的原位制备方法及应用,将碳纳米管进行氨基化改性,性能良好,但是循环性仅有3次。
4.针对以上所述,研发一种简单的方法合成分散性好和循环性能好的纳米催化剂对于降低催化剂成本并提高甲酸制氢反应效率是非常必要的。
技术实现要素:
5.本发明的目的是要针对在催化过程中金属纳米颗粒容易团聚而导致长期稳定性和回收性差的缺点,提供一种氮掺杂碳纳米管和还原氧化石墨烯的双载体上负载钯镍纳米催化剂的制备方法及应用。该方法的关键是将碳纳米管和尿素在空气气氛下煅烧,使其表面富集氮物质,然后将氮掺杂碳纳米管和氧化石墨烯水溶液混合,通过加入氨基,快速实现对双载体的改性,最后负载上pd和ni元素。本发明得到催化剂在甲酸的制氢反应中具有更高的催化活性和循环稳定性,合成过程和操作简单,并且合成周期短。
6.为达到上述目的,本发明是按照以下技术方案实施的:
7.一种双载体上负载钯镍纳米催化剂的制备方法,包括以下步骤:
8.步骤一、在空气氛围下,将碳纳米管和尿素在230~300℃下煅烧,煅烧时间为1~6h,得到氮掺杂碳纳米管;
9.其中,质量比为碳纳米管:尿素=(1~3):(1~3);
10.步骤二、将氮掺杂碳纳米管(n-cnts)和氧化石墨烯(go)溶液加入到去离子水中,超声处理10~30min,得到混合溶液a;
11.其中,每10~50ml去离子水中加入10~60mg n-cnts和10~60mg go;
12.go水溶液浓度为1~15mg/ml;
13.步骤三、将apts加入到步骤二的混合溶液a中,并继续搅拌10~30min后,得到混合溶液b;
14.其中,每10~20ml混合溶液a中加入0.1~0.8mlapts;
15.步骤四、将na2pdcl4和nicl2溶液加入到混合溶液b中,搅拌10~30min后,得到混合溶液c;
16.其中,摩尔比为na2pdcl4:nicl2=(1~9):1,每10~20ml混合溶液b中加入0.01~0.05mmol的na2pdcl4;
17.步骤五、将nabh4作为还原剂加入到步骤四的混合溶液c中,磁力搅拌还原20~60min,得到混合溶液d;
18.其中,每15~25ml混合溶液c加入20~60mg nabh4;
19.步骤六、在室温下,当步骤五所述的混合溶液d没有气泡时,经过离心、水洗,即可得到氮掺杂碳纳米管和还原氧化石墨烯的双载体上负载钯镍纳米催化剂(pdni/nh
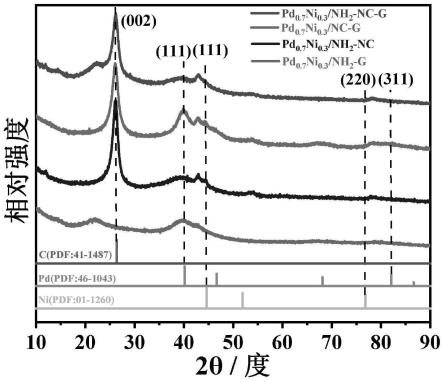
2-nc-g)。
20.所述步骤一中的煅烧方法优选为:先将碳纳米管与50~70%的尿素混合后,在270~300℃下煅烧0.5~2h,洗涤干燥后,加入剩余的尿素,在230~270℃下煅烧0.5~4h;
21.所述pdni为两金属合金结构,且均匀的分散在双载体上,颗粒尺寸为1.0~2.6nm。
22.所述步骤三中apts的纯度为98%。
23.所述步骤四中na2pdcl4和nicl2水溶液的浓度为0.02~0.1m。
24.所述步骤五中的nabh4与混合溶液c进行还原反应的温度为室温。
25.所述步骤六中的离心的转速为8000~12000rpm,时间为3~10min。
26.所述的方法制备的双载体上负载钯镍纳米催化剂的应用,将该催化剂应用于催化甲酸室温分解制氢反应;
27.具体包括如下步骤:将得到的催化剂分散在水中,再加入甲酸水溶液,温度为25~50℃、常压,即可催化甲酸分解制氢;
28.其中,每5~20ml去离子水中加入0.05~1mmol催化剂;甲酸水溶液的浓度为0.1~5m,催化剂的摩尔量以pd和ni两种元素的摩尔量之和计。
29.与现有技术相比,本发明的有益效果是:
30.本发明是一种双载体上负载钯镍纳米催化剂的制备方法及应用,采用了简单的浸渍法合成了平均尺寸为1.8~2.0nm的氨基化改性的氮掺杂碳纳米管(n-cnts)和还原氧化石墨烯(rgo)的复合载体(nh
2-nc-g)负载超细pdni纳米粒子催化剂。值得注意的是,所制备的pd
0.7
ni
0.3
/nh
2-nc-g催化剂表现出良好的甲酸制氢催化活性,在323k时tof值为3899.85h
‑1和ea值为34.67kj mol-1
,这和大多数报道的非贵金属催化剂相当。优化后的pd
0.7
ni
0.3
/nh
2-nc-g催化剂在7次反应中表现出优异的稳定性,100%的转化率和100%的h2选择性。这可以归因于pdni纳米粒子的高分散性,氨基对o-h裂解的促进作用和丰富的活性位点的综合作用。这项工作不仅为甲酸系统在燃料电池上的实际应用提供更多可能而且为其他催化领域的催化剂开发提供了更多的可能性和参考意义。
附图说明
31.图1为实施例1中pdni/nh
2-nc-g催化剂的制备示意图;
32.图2为实施例1、比较例1、比较例2和比较例3制备的催化剂的x射线衍射谱图;
33.图3为实施例1中pdni/nh
2-nc-g催化剂的x射线光电子能谱图;其中,图3(a)为pd 3d的x射线光电子能谱图,图3(b)为ni 2p的x射线光电子能谱图;
34.图4为实施例1中pdni/nh
2-nc-g催化剂的透射电子显微镜图片;
35.图5为实施例1、比较例1、比较例2和比较例3制备的催化剂在50℃下催化甲酸分解的时间-过程曲线;
36.图6为实施例1中pdni/nh
2-nc-g催化剂在50℃下催化甲酸分解的循环性能曲线图。
具体实施方式
37.下面结合具体实施例对本发明作进一步描述,在此发明的示意性实施例以及说明用来解释本发明,但并不作为对本发明的限定。
38.实施例1
39.1.如图1所示,一种双载体上负载钯镍纳米催化剂的制备方法,包括以下步骤:
40.将0.2g碳纳米管和0.3g尿素混合均匀后放入瓷舟中,然后在300℃的空气气氛下保温2h。待混合物冷却至室温后,用去离子水离心洗涤三次,然后在60℃下真空干燥过夜(得到0.2g样品)。将得到的0.2g煅烧好的样品再次和0.1g尿素混合,在250℃的空气气氛下保温4h。待冷却至室温后,用去离子水离心洗涤三次,然后在60℃下真空干燥过夜。命名为n-cnts。
41.将3.36ml go水溶液(8.92mg/ml)和40mg n-cnts分散在10ml去离子水中。在室温下磁力搅拌5min并超声处理15min后得到均匀的黑色分散液。向分散液中加入0.4ml apts,然后磁力搅拌10min得到黑色混合液。向上述混合液中添加na2pdcl4(0.02m,1.75ml)和nicl2(0.02m,0.75ml)溶液,并且磁力搅拌10min。最后,加入30mg nabh4并搅拌20min至不再有气泡产生。将黑色悬浮液高速离心(10000rpm)、水洗三次即可得到pdni/nh
2-nc-g催化剂。
42.2.样品检测
43.(1)将前述制得的pdni/nh
2-nc-g催化剂冷冻干燥;参考图2,x射线粉末衍射(xrd)结果表明,该实验方法成功的合成了氮掺杂碳纳米管和还原氧化石墨烯的双载体上负载钯镍纳米催化剂,且pdni纳米粒子是合金结构。
44.(2)将前述制得的pdni/nh
2-nc-g催化剂在n2气氛下干燥;参考图3,x射线光电子能谱(xps)结果显示,pd和ni之间发生了电子转移。
45.(3)将前述制得的pdni/nh
2-nc-g催化剂稀释,滴在碳支持膜上,干燥;参考图4,透射电子显微镜(tem)结果显示,pdni/nh
2-nc-g样品具有较小的颗粒尺寸(1.0~2.6nm)和均匀的分散性。
46.3.催化甲酸制氢反应
47.将前述全部制得的pdni/nh
2-nc-g催化剂(催化剂的摩尔量以pd和ni两种元素的摩尔量之和计,即催化剂为0.05mmol)分散到10ml去离子水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次pdni/nh
2-nc-g催化剂催化甲酸水溶液制氢过程的气体量(ml)与时间(min)图如图5所示,在50℃下催化甲酸分解制氢2.33min内产生气体的量为245ml,转换率达到100%。
48.在第一轮分解反应结束以后,再将等量的甲酸溶液加入到双颈烧瓶中,剩下的操作与之前反应的相同。将同样的操作步骤在50℃的水浴温度下再重复六次,如图6所示,所制备的pdni/nh
2-nc-g催化剂循环七次的时间分别为2.33、5.00、7.42、10.22、12.40、16.33和25.98min,虽反应速率略有下降,但是仍能提供100%的转化率。经气相色谱(gc)检测仍能提供100%的h2选择性。
49.实施例2
50.其他步骤同实施例1,不同之处为n-cnts的添加量由40mg替换为60mg,在50℃下催化甲酸分解制氢,3.08min内产生气体的量为245ml,转换率达到100%。
51.实施例3
52.其他步骤同实施例1,不同之处为apts的添加量由0.4ml替换为0.8ml,在50℃下催化甲酸分解制氢,3.07min内产生气体的量为245ml,转换率达到100%。
53.实施例4
54.其他步骤同实施例1,不同之处为na2pdcl4的添加量由1.75ml替换为1.25ml,nicl2的添加量由0.75ml替换为1.25ml,在50℃下催化甲酸分解制氢,3.67min内产生气体的量为245ml,转换率达到100%。
55.实施例5
56.其他步骤同实施例1,不同之处为n-cnts的煅烧条件由“300℃煅烧2h,经过离心干燥后,再250℃煅烧4h”替换为“300℃煅烧2h,然后250℃继续煅烧4h”。在50℃下催化甲酸分解制氢,2.45min内产生气体的量为245ml,转换率达到100%。经xps表征显示,n-cnts的氮含量原子百分比由18.07%降低为10.49%。
57.实施例6
58.其他步骤同实施例1,不同之处为n-cnts的煅烧条件由“300℃煅烧2h,经过离心干燥后,再250℃煅烧4h”替换为300℃煅烧6h。在50℃下催化甲酸分解制氢,2.46min内产生气体的量为245ml,转换率达到100%。经xps表征显示,n-cnts的氮含量原子百分比为8.84%。
59.实施例7
60.其他步骤同实施例1,不同之处为n-cnts的煅烧条件由“300℃煅烧2h,经过离心干燥后,再250℃煅烧4h”替换为250℃煅烧6h。在50℃下催化甲酸分解制氢,4.40min内产生气体的量为245ml,转换率达到100%。经xps表征显示,n-cnts的氮含量原子百分比为7.70%。
61.比较例1(未添加go)
62.将40mgn-cnts分散在10ml去离子水中。在室温下磁力搅拌5min并超声处理15min
后得到均匀的黑色分散液。向分散液中加入0.4ml apts,然后磁力搅拌10min得到黑色混合液。向上述混合液中添加na2pdcl4(0.02m,1.75ml)和nicl2(0.02m,0.75ml)溶液,并且磁力搅拌10min。最后,加入30mg nabh4并搅拌20min至不再有气泡产生。将黑色悬浮液高速离心、水洗三次即可得到pdni/nh
2-nc催化剂。
63.将pdni/nh
2-nc催化剂分散到10ml去离子水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次pdni/nh
2-nc催化剂催化甲酸制氢过程的气体量(ml)与时间(min)图如图5所示,在50℃下催化甲酸分解制氢,7.42min内产生气体的量为245ml,转换率达到100%。
64.比较例2(未添加n-cnts)
65.将3.36ml go水溶液(8.92mg/ml)分散在10ml去离子水中。在室温下磁力搅拌5min并超声处理15min后得到均匀的黑色分散液。向分散液中加入0.4ml apts,然后磁力搅拌10min得到黑色混合液。向上述混合液中添加na2pdcl4(0.02m,1.75ml)和nicl2(0.02m,0.75ml)溶液,并且磁力搅拌10min。最后,加入30mg nabh4并搅拌20min至不再有气泡产生。将黑色悬浮液高速离心、水洗三次即可得到pdni/nh
2-g催化剂。
66.将pdni/nh
2-g催化剂分散到10ml去离子水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次pdni/nh
2-g催化剂催化甲酸制氢过程的气体量(ml)与时间(min)图如图5所示,在50℃下催化甲酸分解制氢,4.72min内产生气体的量为245ml,转换率达到100%。
67.比较例3(未添加apts)
68.将3.36ml go水溶液(8.92mg/ml)和40mg n-cnts分散在10ml去离子水中。在室温下磁力搅拌5min并超声处理15min后得到均匀的黑色分散液。向上述混合液中添加na2pdcl4(0.02m,1.75ml)和nicl2(0.02m,0.75ml)溶液,并且磁力搅拌10min。最后,加入30mg nabh4并搅拌20min至不再有气泡产生。将黑色悬浮液高速离心、水洗三次即可得到pdni/nc-g催化剂。
69.将pdni/nc-g催化剂分散到10ml去离子水中,再加入5mmol(1m)的甲酸水溶液,并通过气体量管测量所产生的氢气。此次pdni/nc-g催化剂催化甲酸制氢过程的气体量(ml)与时间(min)图如图5所示,在50℃下催化甲酸分解制氢,30.05min内产生气体的量仅为51ml,转换率为20.82%。
70.通过以上实施例和比较例的对比,pdni/nh
2-nc-g催化剂性能优异的原因可能是以下几点:首先,碳纳米管和尿素煅烧后使碳纳米管表面富集含氮物质,增强了金属纳米颗粒和载体的亲和力。第二,go和n-cnts的大表面积的特点可以提高更多的电子活性位点。第三,双载体的形成进一步限制和阻止了金属纳米颗粒的生长,有利于形成超细尺寸的pdni金属纳米颗粒。第四,氨基的引入提高了双载体的亲水性。最后,经氨基改性后,催化剂保持了和载体一样的介孔结构,并且比表面积和孔体积均增大,更利于金属纳米颗粒的负载。
71.本发明的技术方案不限于上述具体实施例的限制,凡是根据本发明的技术方案做出的技术变形,均落入本发明的保护范围之内。
72.本发明未尽事宜为公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1