一种基于离子注入技术的微纳颗粒二维图形化吸附工艺的制作方法
本发明属于图形化技术领域,涉及一种新的材料表面图形化技术手段,具体涉及一种基于离子注入技术的微纳颗粒二维图形化吸附工艺。
技术背景
新型微观结构的成功构造如微观图案化的表面对于现代科技的发展有着重要的意义且备受关注。随着材料科学的发展,现代材料的性能不再单纯依赖于材料本身的固有性质,通过可控的方法构筑得到的表面微米或纳米尺度有序结构往往带来令人兴奋的崭新现象和功能。各种现代材料的可控微观有序结构化已经成为其器件化和功能高级化的必要前提。
微纳器件广泛地被应用于电子学,光学,计算机科学等领域,有重要的市场价值和国防价值。广义地讲,微纳器件制造领域的图形化就是在材料表面形成图案的过程和技术,从北宋的活字印刷术,到今天的芯片加工,都属于图形化工艺,对于人类社会的进步有重要意义。狭义地来说,微纳器件制造领域的图形化技术,是指在基材表面形成规则的,并具有一定功能的表面结构的技术。微纳器件通常是指尺度从微米量级至纳米量级的,由线(一维)、薄膜或层(二维)和颗粒点阵(零维)构筑的新型器件。因此,微纳器件的获得需要能控制微纳组成单元选择性组装的能力。
传统的微纳器件制造领域的图形化技术包括光刻模板法,聚焦离子束-化学腐蚀法,激光/等离子体刻蚀法,纳米压印法等。然而,刻蚀技术工艺复杂、成本高昂;聚焦离子束成本高,难以大规模生产应用;纳米压印技术的模板制作昂贵费时;微纳模板工艺难于大规模拓展。离子注入技术因与现有的半导体工艺相兼容,它不但可以帮助实现材料表面的选择性,也可以协助实现材料表面的有序组装,是一种具有巨大发展潜力的材料表面的图形化方法。
虽然离子注入工艺广泛地用于半导体掺杂,改善材料表面耐蚀性和硬度,但是离子注入通常会引起材料表面多个物理量的变化,如化学电位、表面电势、表面能等,并且现阶段对离子注入工艺引起材料表面的物理化学性质变化缺乏统一的认识。因此本发明基于离子注入表面zeta电位的变化,提供了一种既简单又稳定,且可大面积加工的微纳颗粒的图形化吸附工艺,为科学研究和工艺设计提供了一种新的方法。
技术实现要素:
有鉴于此,本发明提供一种基于离子注入技术的微纳颗粒二维图形化吸附工艺,作为一种新的灵活的图形化手段,由于具有方法简单、可重复性强的特点,使其在电子学、光学、电池微结构加工和新型计算机等领域中具有广阔的应用前景。
为了实现上述目的,本发明提供如下技术方案:
一种基于离子注入技术的微纳颗粒二维图形化吸附工艺,通过在材料表面覆盖掩膜,注入不同种类的离子,并调节材料表面的zeta电位,在合适的吸附组装环境中,胶体溶液中的不同种类微/纳米颗粒在材料表面实现正/反图形化吸附,所述吸附工艺具体包括如下:
步骤一:在盖有掩膜的目标材料表面注入适当剂量、能量且不同种类离子;
步骤二:室温条件下通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在待吸附的胶体颗粒溶液中垂直静置0.5~60min,之后匀速提拉、平放,经过液相蒸发过程,胶体颗粒最终在材料表面形成本发明公开的微纳颗粒二维吸附图形。
通过采用上述技术方案,本发明的有益效果如下:
本发明基于对离子束与材料表面改性的作用,通过在材料表面覆盖掩膜,并在此基础上注入特定离子,继而选择性地在材料表面形成图形化的离子注入区。并调节合适的吸附组装环境,将材料浸入颗粒胶体溶液中静置,经过提拉平放于桌面自然干燥后,在静电作用下实现微纳尺度的不同种类颗粒的正/反图形化自组装。此外,基于对图形化形成过程的规律探索,通过改变颗粒胶体溶液的张力参数,实现了不同尺度从均匀图案到点阵的图形化工作。
优选的,所述掩膜是依据需求的图形化要求而选取的物理掩膜。
需要说明的是,本发明依据所需求的图形化要求而选择相应孔目数的掩膜。
优选的,所述材料包括有机高分子、半导体、金属或无机陶瓷材料。
优选的,所述注入离子为金属离子(cu+/mo+/fe+/ni+/w+等)、官能团离子(-nh+/-oh+/-coo-等)或部分非金属离子。
其中,金属离子、非金属离子注入具有离子引出源成本较低的优势。在不产生表面形貌较大破坏的情况下,适宜能量的离子的注入会引起材料表面旧化学键的断裂和表面成分相应变化,涉及新相的形成等,进而影响材料表面zeta电位的变化,利用注入区在溶液中静电作用的变化实现图形化吸附。
而官能团离子的注入能够具有目的性地在基体表面修饰官能团,通过与目标颗粒表面基团的化学键或者静电相互作用实现选择性吸附。
优选的,所述离子注入能量选为5~80kev,注入剂量选为1014~1017ions/cm2。
优选的,所述胶体颗粒溶液的浓度为0.1~10g/l;且所述胶体颗粒包括au颗粒、ag颗粒、sio2颗粒、al2o3颗粒、cr(oh)3颗粒、普鲁士蓝纳米颗粒等。
优选的,所述胶体颗粒溶液中溶剂选用乙醇为主(10%~99%)和kcl溶液组成的混合剂,其中kcl溶液可替换为其他离子溶液,且根据不同的吸附情形将所述胶体颗粒溶液的ph值控制在2~12之间。
优选的,所述kcl溶液或其他离子溶液占所述混合剂的体积分数为0.1%-95%;且所述kcl溶液或其他离子溶液的浓度为5~150mg/l。
值得说明的是,经过液相蒸发过程,颗粒与基体表面不同区域的作用力之差成为其分散在特定的区域的驱动力,因而形成吸附图形。并且随着电解质溶液体积分数越高,液相张力越大,其所形成的图案趋于点阵。
经由上述的技术方案可知,与现有技术相比,本发明提供的一种基于离子注入技术的微纳颗粒二维图形化吸附工艺,通过利用离子注入表面zeta的变化而得到一种既简单又稳定,且可大面积加工的微纳颗粒的图形化吸附工艺。本发明单纯依靠材料固有的表面电化学特性,实现灵活自由的图形化吸附,过程简单易行,不仅避免了激光刻蚀、化学腐蚀等方法的繁琐,而且相较聚焦离子注入,成本更低廉;并且掩膜可重复循环利用,适合大面积制备图形化微纳加工。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
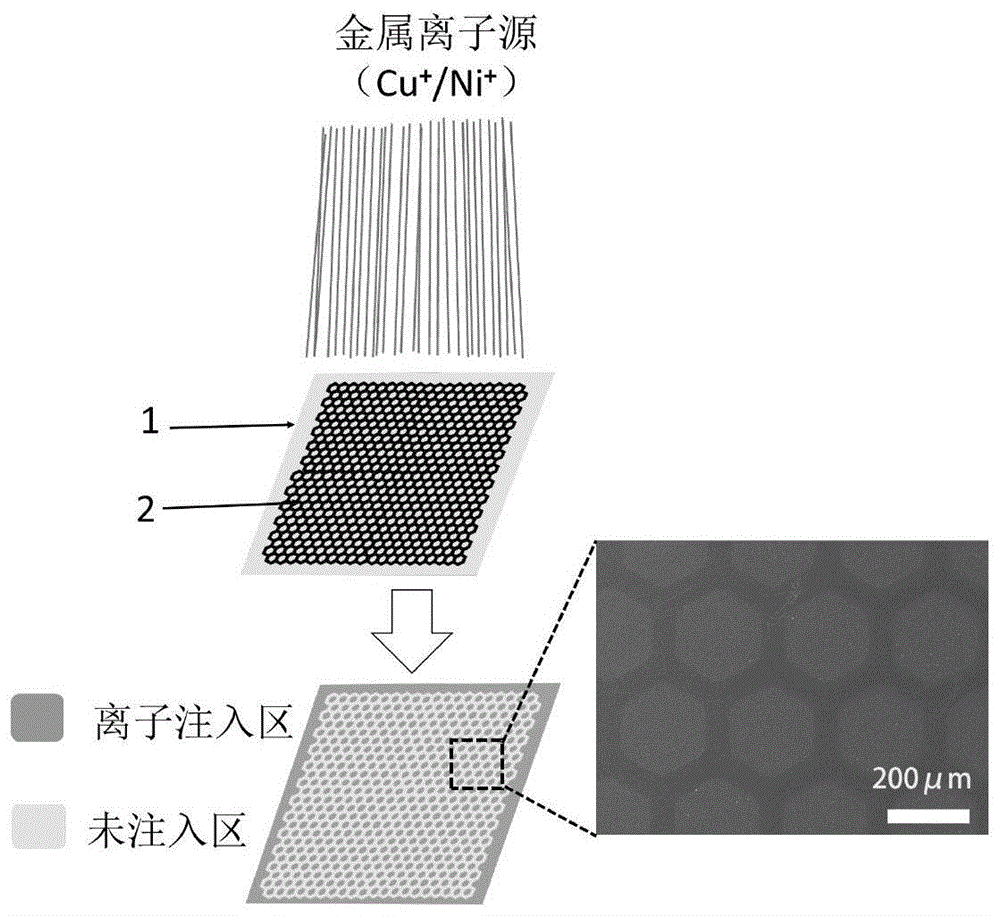
图1为本发明金属掩膜离子注入示意图及注入后pi膜sem图;
图2为本发明离子注入对聚酰亚胺薄膜zeta电位的影响;其中图2(a)为注入cu+或ni+金属离子的pi薄膜材料在胶体溶液中的zeta电位测试结果;图2(b)为金属离子注入后pi薄膜材料在水溶液中的zeta电位测试结果;
图3为本发明胶体颗粒吸附装置示意图;
图4为不同种类的微纳颗粒在聚酰亚胺薄膜材料上的吸附sem图;其中图4(a)为氨基化后的sio2亚微米颗粒吸附于cu+离子注入聚酰亚胺表面;图4(b)为未氨基化的sio2亚微米颗粒吸附于cu+离子注入聚酰亚胺表面;图4(c)为氨基化后的sio2亚微米颗粒吸附于ni+离子注入聚酰亚胺表面;图4(d)为未氨基化的sio2亚微米颗粒吸附于ni+离子注入聚酰亚胺表面;
图5为不同体积百分比的混合吸附液条件下的微纳颗粒在聚酰亚胺薄膜材料上的吸附sem图;其中图5(a)为乙醇与kcl溶液的体积比为9:1;图5(b)为乙醇与kcl溶液的体积比为7:3;图5(c)为乙醇与kcl溶液的体积比为5:5;图5(d)为乙醇与kcl溶液的体积比为3:7;图5(e)为氨基化sio2纳米球吸附于cu+离子注入聚酰亚胺表面;
图6为fe+/cu+离子注入(注入剂量为5*1016ions/cm2,能量为30kev)pi薄膜材料的afm图;表面起伏约为20nm。
图7为10%的cr(oh)3颗粒胶体溶液在fe+注入的聚酰亚胺薄膜材料上的正吸附sem图;
图8为10%普鲁士蓝(fe4[fe(cn)6]3)纳米颗粒(颗粒表面zeta电位为-49.7mv,3-15nm)的正反吸附sem图;其中图8(a)为普鲁士蓝纳米颗粒在n+i离子注入聚酰亚胺薄膜的图形化吸附;图8(b)为普鲁士蓝纳米颗粒在cu+离子注入聚酰亚胺薄膜的图形化吸附。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明实施例公开了一种既简单又稳定,且可大面积加工的微纳颗粒二维图形化吸附工艺。
为更好地理解本发明,下面通过以下实施例对本发明作进一步具体的阐述,但不可理解为对本发明的限定,对于本领域的技术人员根据上述发明内容所作的一些非本质的改进与调整,也视为落在本发明的保护范围内。其中,所述聚酰亚胺薄膜是一种耐热、绝缘、高强度的高分子材料,被广泛应用于生产、生活各个领域。
需要说明的是,本发明的使用范围不局限于聚酰亚胺等有机高分子薄膜材料,同样也适用于半导体,金属和无机陶瓷材料。
一种基于离子注入技术的微纳颗粒二维图形化吸附工艺,所述吸附工艺具体包括如下:
步骤一:在盖有掩膜的材料表面注入不同种类的离子;
步骤二:室温条件下通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在待吸附的胶体颗粒溶液中垂直静置0.5~60min,之后匀速提拉、平放,经过液相蒸发过程,胶体颗粒最终在材料表面形成本发明公开的微纳颗粒二维吸附图形。
为了进一步实现本发明的技术效果,所述材料包括有机高分子、半导体、金属或无机陶瓷材料。
为了进一步实现本发明的技术效果,所述掩膜是依据需求的图形化要求而选取的物理掩膜,掩膜材质包括纯金属,高分子或其他无机化合物。
为了进一步实现本发明的技术效果,所述离子包括金属离子、非金属离子或官能团离子。
为了进一步实现本发明的技术效果,所述离子注入能量选为5~80kev,注入剂量选为1014~1017ions/cm2。
为了进一步实现本发明的技术效果,所述胶体颗粒溶液的浓度为0.1~10g/l;且所述胶体颗粒包括sio2颗粒、al2o3颗粒、cr(oh)3颗粒或普鲁士蓝纳米颗粒。
为了进一步实现本发明的技术效果,所述胶体颗粒溶液中溶剂选用乙醇和kcl溶液或其他离子溶液组成的混合剂,且将所述胶体颗粒溶液的ph值控制在2~12之间。
为了进一步实现本发明的技术效果,所述kcl溶液或其他离子溶液占所述混合剂的体积分数为0.1%-95%。
下面,将结合具体实施例,对本发明的技术方案进行进一步的说明。
1.改变吸附颗粒种类
实施例1:
一种基于离子注入技术的微纳颗粒二维图形化吸附工艺,具体包括如下:
步骤一:在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入cu+金属离子,其中注入能量选为30kev,注入剂量选为5×1016ions/cm2;
步骤二:室温条件下(25℃)通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在sio2颗粒溶液中垂直静置3min,之后以1mm/s的速度提拉,平放一段时间,经过液相蒸发过程,颗粒与基体表面不同区域的作用力之差成为其分散在特定的区域的驱动力,从而在材料表面形成本发明公开的微纳颗粒二维负吸附图形。
实施例2:
步骤一:在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入ni+金属离子,其中注入能量选为30kev,注入剂量选为5×1016ions/cm2;
步骤二:室温条件下(25℃)通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在sio2颗粒溶液中垂直静置3min,之后以1mm/s的速度提拉,平放一段时间,经过液相蒸发过程,颗粒与基体表面不同区域的作用力之差成为其分散在特定的区域的驱动力,从而在材料表面形成本发明公开的微纳颗粒二维正吸附图形。
实施例3:
本实施例与实施例1存在的区别为:本实施例选用的胶体颗粒溶液为氨基化的sio2颗粒溶液,其他工艺步骤与参数不变。从而在材料表面形成与本发明公开的实施例1和实施例2中的图形相反的吸附图形。
实施例4:
本实施例与实施例1存在的区别为:本实施例选用的胶体颗粒溶液为cr(oh)3颗粒溶液,其他工艺步骤与参数不变。
实施例5:
本实施例与实施例3存在的区别为:本实施例选用的胶体颗粒溶液为cr(oh)3颗粒溶液,注入离子种类为fe+。其他工艺步骤与参数不变。
实施例6:
本实施例与实施例1存在的区别为:本实施例选用的胶体颗粒溶液为普鲁士蓝纳米颗粒溶液,其他工艺步骤与参数不变。
2.改变混合吸附液的体积百分比
实施例7:
一种基于离子注入技术的液相张力作用影响的微纳颗粒二维图形化吸附工艺,具体包括如下:
步骤一:在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入cu+或ni+金属离子,其中注入能量选为30kev,注入剂量选为5×1016ions/cm2;
步骤二:室温条件下(25℃)配置乙醇%vol:kcl溶液%vol为9:1的吸附溶液,其溶液张力为25.0mn/m;通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在sio2/氨基化sio2颗粒溶液中垂直静置3min,之后以1mm/s的速度提拉,平放一段时间,在液相蒸发过程中,不同的液相张力的不同大小的拖拽作用使得其在材料表面形成本发明公开的微纳颗粒二维吸附图形。
实施例8:
一种基于离子注入技术的液相张力作用影响的微纳颗粒二维图形化吸附工艺,具体包括如下:
步骤一:在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入cu+或ni+金属离子,其中注入能量选为30kev,注入剂量选为5×1016ions/cm2;
步骤二:室温条件下(25℃)配置乙醇%vol:kcl溶液%vol为7:3的吸附溶液,其溶液张力为28.1mn/m;通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在sio2/氨基化sio2颗粒溶液中垂直静置3min,之后以1mm/s的速度提拉,平放一段时间,在液相蒸发过程中,不同的液相张力的不同大小的拖拽作用使得其在材料表面形成本发明公开的微纳颗粒二维吸附图形。
实施例9:
一种基于离子注入技术的液相张力作用影响的微纳颗粒二维图形化吸附工艺,具体包括如下:
步骤一:在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入cu+或ni+金属离子,其中注入能量选为30kev,注入剂量选为5×1016ions/cm2;
步骤二:室温条件下(25℃)配置乙醇%vol:kcl溶液%vol为5:5的吸附溶液,其溶液张力为31.8mn/m;通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在sio2/氨基化sio2颗粒溶液中垂直静置3min,之后以1mm/s的速度提拉,平放一段时间,在液相蒸发过程中,不同的液相张力的不同大小的拖拽作用使得其在材料表面形成本发明公开的微纳颗粒二维吸附图形。
实施例10:
一种基于离子注入技术的液相张力作用影响的微纳颗粒二维图形化吸附工艺,具体包括如下:
步骤一:在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入cu+或ni+金属离子,其中注入能量选为30kev,注入剂量选为5×1016ions/cm2;
步骤二:室温条件下(25℃)配置乙醇%vol:kcl溶液%vol为3:7的吸附溶液,其溶液张力为37.9mn/m;通过升降台浸渍提拉的方法,将步骤一选择性注入的材料浸没在sio2/氨基化sio2颗粒溶液中垂直静置3min,之后以1mm/s的速度提拉,平放一段时间,在液相蒸发过程中,不同的液相张力的不同大小的拖拽作用使得其在材料表面形成本发明公开的微纳颗粒二维吸附图形。
下面将结合本发明公开的说明书附图,对本发明的技术方案进行进一步的说明。
请参见说明书附图1,本发明在盖有mo金属掩膜(孔目数为100目,且孔为边长为160μm的六边形)的聚酰亚胺薄膜表面注入cu+或ni+金属离子。
参见说明书附图3,将注入cu+或ni+金属离子的聚酰亚胺薄膜浸没在胶体颗粒溶液中垂直静置3min,之后以1mm/s的速度匀速提拉,平放一段时间,经过液相蒸发过程,胶体颗粒最终在材料表面形成本发明公开的微纳颗粒二维吸附图形,如附图4所示。
注入cu+或ni+金属离子的pi薄膜材料浸没在胶体颗粒溶液中的zeta电位测试结果参见附图2,经测量,聚酰亚胺在胶体颗粒溶液中的表面zeta电位为-7.6mv,经过cu+离子注入,其电位升高至-0.9mv,而ni+离子注入,其电位变为-16.7mv。可见,对于聚酰亚胺薄膜来说,cu+离子和ni+离子对于pi膜的电位有着完全相反的影响。由于cu+、ni+离子的注入会在高分子薄膜基体表面形成相应的金属氧化物及单质,同时伴随有化学键的断裂,因而会造成材料表面zeta电位的改变,具体说来,cu+离子的注入使得聚酰亚胺基体表面zeta电位向正值改变,ni+离子注入使得电位向更负值改变。
对此,我们提出公式:
k=lnr
来衡量注入金属离子前后表面电位的变化情况,其中r为注入离子后pi电位/pi电位。k值越大,反映了注入前后电位变化情况越明显,相应图形化效果更好。
另外,发明人还研究了不同种类的微纳颗粒在聚酰亚胺材料上的吸附情况,参见附图4。其中图4a和图4b为cu+离子注入;图4c和图4d为ni+离子注入;并且图4a和图4c为氨基化后的sio2亚微米颗粒(zeta电位为+42.3mv);图4b和图4d为未氨基化的sio2亚微米颗粒(zeta电位为-40.1mv)。
由图4a和图4c得知,对于氨基化的sio2颗粒来说,经实验测得其表面zeta电位为正值(+42.3mv),在吸附过程中倾向于吸附在电位表现更负的位置,这一位置在cu+离子注入的情况下为未注入区,在ni+离子注入的情况下为注入区。
而图4b和图4d得知,对于未氨基化的sio2颗粒来说,其表面zeta电位为负值(-40.1mv),此时无论注入何种离子,都表现为斥力,但是不同注入离子引起的负值的相对大小会使得同为负电位的亚微米颗粒积累在绝对值较小的负电位区域,同样也能实现图形化,其图案与氨基化颗粒所形成的图案相反。
对此我们提出公式:
来对吸附规律进行总结。式中
并且通过离子注入改变pi膜表面的zeta电位,可以获得针对不同表面zeta电位颗粒的不同选择性图形化吸附。
其中,金属离子注入后,注入区与未注入区的afm图参见附图6。从图6中可以看出金属离子注入后,会在pi膜上形成深度约为20nm的凹陷区域(注入区)。
此外,吸附位置确定以后,改变其中kcl的体积分数为10%,30%,50%,70%。体积分数越高,液相张力越大,形成图案趋于点阵。具体参见附图5。图5a-图5d分别为在10%,30%,50%,70%的kcl溶液(vol%)中的sio2颗粒在聚酰亚胺材料上的吸附sem图。图5e为70%vol的kcl溶液的混合吸附液条件下的氨基化的sio2颗粒在聚酰亚胺材料上的吸附sem图。
由图5可见,随着乙醇体积分数的减小,液相表面的张力增加,液相在蒸发过程中拖拽力增加,即便颗粒有存在特定区域的趋势,但经过蒸发后会被大的表面张力拖拽收缩,最终形成点阵列。
另外,发明人分别对含90%vol乙醇的cr(oh)3颗粒和普鲁士蓝(fe4[fe(cn)6]3)纳米颗粒分散液在聚酰亚胺材料上的正反吸附情况进行研究,具体结果参见附图7和附图8。图7反映了表面zeta电位为正值的cr(oh)3吸附在fe+离子注入(基体zeta电位变低)的材料表面。图8反映了表面zeta电位为负值的普鲁士蓝颗粒分别吸附在ni+离子和cu+离子注入的pi表面。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!