一种N-GO@Co-Ni
一种n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料及其制备方法
技术领域
1.本发明属于电化学电极材料与技术领域,涉及n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料及其制备方法,具体地说,是涉及一种多功能电解用原位生长于泡沫镍钴(ncf)金属集流体上的氮掺杂氧化石墨烯(n
‑
go)包覆的co掺杂ni
12
p5‑
ni3p复合电极材料及其制备方法
背景技术:
2.h2作为绿色能源载体,有望在未来发挥重要作用。电催化水分解作为一种提供h2的环保方法引起了全世界的兴趣。目前,人们对电催化分解水的析氢反应(her)和析氧反应(oer)进行了大量的研究工作。在已公开的技术中,钴和镍等过渡金属的磷化物、氮化物和硫化物已被证明是优异的her电催化剂,一些过渡金属氧化物和双金属氢氧化物显示出优异的oer性能。在上述电催化剂中,过渡金属磷化镍正在成为一种有前途的电催化剂,主要是由于它们的金属特性、优异的导电性以及丰富的化合价,此外,磷的表面极化使磷的中心带负电加速了电子的快速传输并加速了氧化还原反应。然而,其性能仍不足以替代贵金属基催化剂,因此需要更合理的设计以获得优越的性能。
3.另外,阳极的oer仍然是全解水产氢的难点,因为需要更高的过电位来匹配her的速率。同时,oer的产物o2没有显着价值,并且与h2混合后,去除耗能耗时。这种情况下,生物质氧化反应由于其低过电位和高商业价值而引起人们的注意。5
‑
羟甲基糠醛(hmf)被认为是研究最广泛的生物质衍生模型分子之一,并可以进一步氧化成多种高附加值的复合中间体,如2,5
‑
呋喃二甲酸(fdca)等。在高分子材料领域,fdca可以替代对苯二甲酸生产可生物降解且性能良好的生物基聚酯。将hmf氧化为fdca的传统多相催化体系,通常需要使用贵金属基催化剂(例如au、pt、ru和pd),在高温(30
‑
130℃)和高压空气或o2下(例如,0.3
‑
2.0mpa)进行反应。通过电化学方法将hmf氧化制备fdca,可以在环境温度下进行,避免使用高压o2或其他危险的化学氧化剂,对环境友好,但贵金属基电催化剂仍然是最常用的催化剂。因此,高性能非贵金属电催化剂的开发研究是电催化反应工业化应用的关键。然而,目前高性能的磷化镍基复合电催化剂制备仍存在制备工艺过程繁琐,电催化活性低和循环稳定性不足的缺点,因此需要进一步探索开发制备工艺简单,性能优良的高效、稳定和耐用的磷化镍基复合电极材料。本发明精心设计了新型的三组分低共熔混合物液体作为前驱体,将该液体涂覆在预处理过的泡沫镍钴金属片上,经铝箔包裹,在氮气氛下通过简单的一步煅烧,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
技术实现要素:
4.本发明针对现有技术中制备磷化镍基复合电极材料过程复杂繁琐、需要多步合成,难以实现紧密的界面耦合,活性位点低,电催化效率低,长期稳定性差等缺点,提出了一种n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料及其制备方法,其特征在于,氮掺杂氧化石墨烯(n
‑
go)包覆的co掺杂ni
12
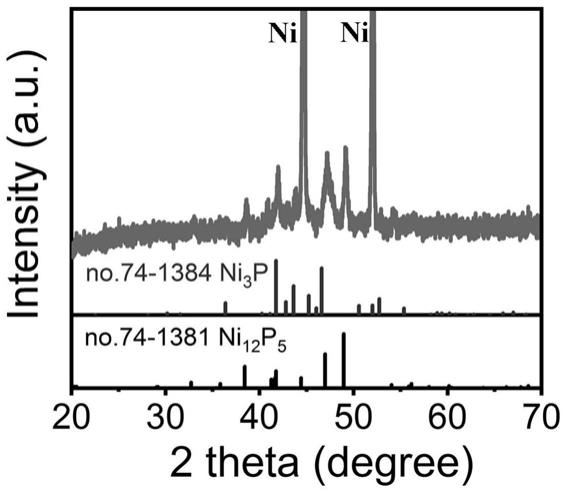
p5‑
ni3p复合磷化物纳米棒组装体,原位生长于泡沫镍钴(ncf)金属片集流体上,所述复合磷化物纳米棒之间相互连接生长形成片状网络结构,由涂覆在预
处理过的泡沫镍钴金属片上的液体前驱体,在铝箔包裹和氮气氛保护下,热解、碳化和磷化反应一步完成,具体包括下述步骤:
5.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
6.(2)将0.001
‑
0.1mol甲氧基甲基三苯基氯化膦、0.001
‑
0.05mol尿素和0.001
‑
0.05mol乙二醇混合,30
‑
90℃搅拌形成透明液体;
7.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到300
‑
650℃,保温1
‑
5h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
8.本发明的优点在于:设计的新型三组分低共熔混合物液体作为前驱体,可以使前驱体碳化、磷化与氧化石墨烯壳层的生长和复合一步完成;co
‑
ni
12
p5‑
ni3p表面的氧化石墨烯壳层可以有效防止催化剂失活,原位生长于泡沫镍钴金属片上的co
‑
ni
12
p5‑
ni3p催化剂,直接用作电极,有效减少论文催化剂的脱落、提高了稳定性;通过co掺杂进行ni
12
p5‑
ni3p催化剂的电子结构调节,可以进一步提高ni
12
p5‑
ni3p的电催化活性;合成的电催化剂是一种多功能电催化剂,可以进行5
‑
羟甲基糠醛(hmf)的电催化氧化、对于电催化分解水制氢和电化学降解有机染料都有高效的电催化活性和稳定性。
附图说明
9.图1为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的xrd谱图。
10.图2为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的拉曼图谱(a)和红外光谱(b)。
11.图3为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料不同放大倍数的sem照片。
12.图4为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的tem照片(a)和hrtem(b)照片。
13.图5为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的stem照片和stem
‑
mapping元素分布图。
14.图6为利用本发明实施例一、对比例一、对比例二所述方法制备的复合电极材料的电催化析氢速率图。
15.图7为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料用于oer和电催化氧化hmf制备fdca的催化性能。(a)水oer和hmf氧化的lsv曲线,(b)hmf制备fdca的选择性,(c)hmf制备fdca的循环稳定性实验。
具体实施方式
16.下面通过实施例对本发明作进一步详细说明:
17.实施例一:
18.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,
放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
19.(2)将0.02mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.01mol乙二醇混合,80℃搅拌形成透明液体;
20.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到450℃,保温4h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
21.实施例二:
22.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
23.(2)将0.02mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.01mol乙二醇混合,80℃搅拌形成透明液体;
24.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到350℃,保温4h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
25.实施例三:
26.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
27.(2)将0.01mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.02mol乙二醇混合,60℃搅拌形成透明液体;
28.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到450℃,保温4h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
29.实施例四:
30.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
31.(2)将0.02mol甲氧基甲基三苯基氯化膦、0.01mol尿素和0.1mol乙二醇混合,60℃搅拌形成透明液体;
32.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到350℃,保温4h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
33.实施例五:
34.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,用铝箔包裹后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
35.(2)将0.03mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.02mol乙二醇混合,70
℃搅拌形成透明液体;
36.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到550℃,保温2h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
37.实施例六:
38.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,用铝箔包裹后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
39.(2)将0.1mol甲氧基甲基三苯基氯化膦、0.05mol尿素和0.05mol乙二醇混合,80℃搅拌形成透明液体;
40.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到600℃,保温1h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
41.实施例七:
42.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
43.(2)将0.02mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.02mol乙二醇混合,50℃搅拌形成透明液体;
44.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,用铝箔包裹后,在氮气气氛保护下加热到350℃,保温4h,冷却后,得到n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
45.对比例一:
46.(1)泡沫镍(nf)金属片的预处理:将泡沫镍金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
47.(2)将0.02mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.01mol乙二醇混合,80℃搅拌形成透明液体;
48.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍上,用铝箔包裹后,在氮气气氛保护下加热到450℃,保温4h,冷却后,得到n
‑
go@ni
12
p5‑
ni3p/nf复合电极材料。
49.对比例二:
50.(1)泡沫镍钴金属片的预处理:将泡沫镍钴金属片剪成1cm
×
1cm大小的正方形片,放入丙酮中超声浸泡5min,再放入1mol/l的稀盐酸中超声浸泡5min,然后,将泡沫镍钴取出分别用去离子水和无水乙醇超声洗涤三次,在40℃的真空干燥箱中干燥;
51.(2)将0.02mol甲氧基甲基三苯基氯化膦、0.005mol尿素和0.01mol乙二醇混合,80℃搅拌形成透明液体;
52.(3)将步骤(2)所得透明液体涂覆在步骤(1)预处理的泡沫镍钴上,在氮气气氛保护下加热到450℃,保温4h,冷却后,得到c@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料。
53.图1为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材
料的xrd谱图。由图可以看出,除了金属镍的衍射峰外,所有衍射峰分别对应于ni3p(jpcds74
‑
1384)和ni
12
p5(jpcds 74
‑
1381)的衍射峰。说明所制备产物为磷化物复合材料。
54.图2为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的拉曼图谱(a)和红外光谱(b)。由拉曼图谱可以看出,样品在1346cm
‑1和1585cm
‑1处有两个典型的特征峰,分别与石墨结构碳的d峰和g峰匹配,d峰和g峰的强度比值i
d
/i
g
=0.84,说明所得产物石墨化程度不完全,缺陷较多。红外光谱中的~3400cm
‑1吸收峰与碳上的
‑
oh伸缩振动相对应,2920cm
‑1和2850cm
‑1处的吸收对应于ch或ch2的烷基伸缩振动,1630cm
‑1处的峰为c=c或c
‑
c的骨架振动峰,表明样品上存在类石墨烯结构,~1717cm
‑1和~1130cm
‑1处的峰来自于c=o和c
‑
o的振动。红外光谱进一步表明样品中石墨烯结构的碳含有大量的
‑
oh,c=o和c
‑
o,是氧化石墨烯结构的碳。
55.图3为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料不同放大倍数的sem照片。从图3a可以看出泡沫镍钴金属片的三维多孔道开放骨架结构,骨架表面均匀的包覆了一层材料,从局部放大照片图3b可以看出,泡沫镍钴骨架表面原位生长了一层类似纳米片的阵列或组装结构,进一步放大后,从图3c可以看出,这些纳米片实际上是由纳米棒之间相互连接生长形成的片状网络结构,复合磷化镍片的网络结构和泡沫镍钴金属片的多孔道开放骨架结构可以使催化剂暴露更多的活性位点,提供快速的导电通道,加快电子/电荷转移速率,从而大大提高电催化活性和稳定性。
56.图4为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的tem照片(a)和hrtem(b,c)照片。从图4a可以看出,在泡沫镍钴的骨架上原位生长的纳米棒相互搭接而成的片状网络结构,单根纳米棒直径约25nm。图4b是单根纳米棒的hrtem照片,可以看出,纳米棒外面包覆一层go碳层,纳米棒晶格明显,从放大的区域图4c可以看出,纳米棒磷化物呈现出多种晶格条纹,也可以清楚的观察到莫尔条纹,说明这些纳米棒是由多层纳米片叠加而成,在纳米棒的边缘的晶面间距是0.200nm和0.224nm,对应于ni3p的(240)和(400)晶面,在纳米棒内部的晶面间距是0.274nm,对应于ni
12
p5的(130)晶面,进一步说明复合电极由go包覆的ni
12
p5‑
ni3p复合材料构成。
57.图5为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的stem照片和stem
‑
mapping元素分布图。stem照片更加清晰地展示了纳米棒相互搭接而成的片状网络结构,从元素分布看,ni、p元素的元素分布最致密,说明纳米棒主要是复合磷化镍,这也与xrd结果一致,co元素均匀分布,结合xrd结果,说明co存在于复合磷化镍的晶格中,没有独立co的磷化物晶相。碳元素分布也很明显,但显示的纳米棒较粗,说明c包覆于复合磷化镍纳米棒表面,这也与hrtem结果一致。同样,o、n元素分布较弱,显示的纳米棒也较粗,说明o、n元素主要存在于纳米棒表面的碳层中,进一步说明是n掺杂的氧化石墨烯,电极材料是n
‑
go@co
‑
ni
12
p5‑
ni3p复合材料。
58.图6为利用本发明实施例一、对比例一、对比例二所述方法制备的复合电极材料在1mol l
‑1koh溶液中的电催化析氢速率图。从图中可看出,实施例一复合电极材料在10ma cm
‑2电流下的过电位是53.8mv,远低于对比例一、对比例二复合电极材料在10ma cm
‑2电流下的过电位212mv和235mv,说明本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料的her活性远高于对比例一、对比例二复合电极材料,co元素对复合磷化物的掺杂和n
‑
go对复合磷化物的包覆能够大大地提高复合磷化物的电催化活性。
59.图7为利用本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料用于oer和电催化氧化hmf制备fdca的催化性能。(a)水oer和hmf氧化的lsv曲线,(b)hmf制备fdca的选择性,(c)hmf制备fdca的循环稳定性实验。由图7a实施例一复合电极材料用于oer和电催化氧化hmf制备fdca的lsv曲线,可以看出,1mol l
‑1koh水溶液oer反应达到20ma/cm2电流需要1.57v过电位,而对于电催化氧化hmf制备fdca,20ma/cm2电流下过电位降低到1.37v,说明碱性条件下,实施例一复合电极材料用于电催化氧化hmf制备fdca性能优于oer反应。图7b是电催化氧化hmf制备fdca过程中,采用高效液相色谱分析体系中hmf和各种氧化产物的浓度随时间变化情况,可以看出,随着hmf浓度的减小,fdca的产率逐渐升高,120min后fdca的浓度达到最大值,接近100%,而其他的氧化产物的浓度很小且没有明显变化,说明实施例一复合电极材料用于电催化氧化hmf制备fdca具有很高的选择性。图7c显示,该复合电极材料用于电催化氧化hmf制备fdca循环使用6次,fdca的产率没有明显的降低,说明该复合电极材料用于电催化氧化hmf制备fdca具有很高的催化活性和选择性,也具有很高的稳定性。
60.进一步将本发明实施例一所述方法制备的n
‑
go@co
‑
ni
12
p5‑
ni3p/ncf复合电极材料用于对水溶液中常见有机染料进行电催化降解,结果表明本发明所制备的复合电极材料对于常见有机染料的电催化降解也有很好的电催化活性,可以用于有机废水的电催化氧化处理。
61.上述实施例是本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,未背离本发明的原理与工艺过程下所作的其它任何改变、替代、简化等,均为等效的置换,都应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1