一种由γ-羟基胺电化学合成取代吲哚的方法
一种由
γ-羟基胺电化学合成取代吲哚的方法
技术领域
1.本发明属于医药化学与精细化学合成领域,具体涉及一种由γ-羟基胺电化学合成取代吲哚的方法。
背景技术:
2.吲哚类化合物的基本骨架为苯并吡咯环,此类化合物广泛存在于具有活性的生物体、天然产物及药物分子结构中。以吲哚为基本母核结构的化合物具有抗肿瘤、抗炎、抗菌等多种药理活性,因此对于该类化合物的合成与开发应用是方法学研究显得尤为重要,且同时具有广阔的发展前景。
3.通过前期检索发现,和c自由基相比,n自由基还没有在合成化学中得到很多关注,这可能是因为它们的反应性较高,同时有效的制备方法也比较缺乏。大多数情况下,n自由基是通过活性n-x键的断裂而产生的,其中x可以是卤原子或者n、o、s基团。同时,大部分的n自由基的r1r2n-x型前体都不太稳定,需要原位合成,也正因如此开发新的、稳定的、易于制备的、廉价的和有效的n-自由基前体是非常重要的。
4.而含氮化合物的合成中,最重要也是最难完成的环节就是碳氮键的构建。碳氮键的构建反应在有机化学中是一类非常重要的反应,但很多有机反应在构建碳氮键的同时也造成了一定的环境污染问题。随着“绿色化学”呼声的增高,化学家越来越青睐于在绿色、无污染的条件下进行有机反应,而在电化学催化下诱导的的碳氮键构建反应合成出新化合物使其具有一定的生物活性与药物活性,或对已有化合物进行功能性改造与修饰,使其能广泛应用于化学工业及精细化工品的生产。能很好地实现绿色化学的目标。
技术实现要素:
5.发明目的:本发明所要解决的技术问题是针对现有技术的不足,提供一种由γ-羟基胺电化学合成取代吲哚的方法,以解决现有技术存在的反应效率低和金属残留大的问题。
6.为了实现上述目的,本发明采取的技术方案如下:
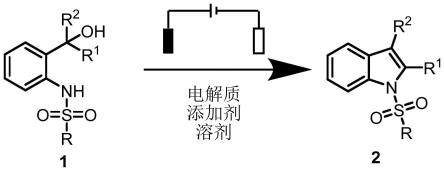
7.一种由γ-羟基胺电化学合成取代吲哚的方法,其特征在于,将底物1、有机添加剂、电解质和溶剂混合得到的均相溶液,加入到具有阴、阳极的电化学反应装置中,通过电催化一步法合成取代吲哚2;
8.反应方程式如下:
9.10.其中,r1选自供电子基团氢、1~4个c的烷基、苯基或取代苯基中的任意一种;
11.r2选自供电子基团氢、1~4个c的烷基、苯基或取代苯基中的任意一种;
12.r选自1~4个c的烷基、苯基或取代苯基中的任意一种。
13.具体地,所述的有机添加剂选自三氟乙酸、甲磺酸、醋酸、对甲苯磺酸中的任意一种或两种以上的组合;优选为醋酸和/或对甲苯磺酸;进一步优选为对甲苯磺酸。
14.具体地,所述的电解质为四丁基六氟磷酸铵、四丁基四氟硼酸铵、四丁基醋酸铵、四丁基碘化铵或高氯酸锂中的任意一种或两种以上的组合;优选为四丁基六氟磷酸铵、四丁基四氟硼酸铵或高氯酸锂中的任意一种或两种以上组合;进一步优选为四丁基六氟磷酸铵和/或四丁基四氟硼酸铵;更进一步优选为四丁基四氟硼酸铵。
15.具体地,所述的溶剂选自二氯乙烷、n,n-二甲基甲酰胺、二甲基亚砜、三氟乙醇、乙腈、六氟异丙醇、三氯乙烯、n,n-二甲基苯胺、四氢呋喃中的任意一种或两种以上的组合;优选为二甲基亚砜、三氟乙醇、乙腈、六氟异丙醇、三氯乙烯、n,n-二甲基苯胺、四氢呋喃中的任意一种或两种以上组合;进一步优选为三氟乙醇、乙腈或六氟异丙醇中的任意一种或两种以上组合;更进一步优选为体积比3.5:6.5的三氟乙醇和乙腈混合溶液。
16.具体地,所述底物1和有机添加剂的摩尔比为1:1~3;优选1:1~2,进一步优选为1:1~1.8,更进一步优选为1:1.5。
17.具体地,所述的底物1和电解质的摩尔比为1:0.5~2;优选为1:0.5~1.5,进一步优选为1:1~1.5,更进一步优选为1:1。
18.具体地,所述的底物1在均相溶液中浓度为0.02~0.08mmol/ml,优选为0.02~0.06mmol/ml,进一步优选为0.02~0.04mmol/ml,更进一步优选为0.03mmol/ml。
19.具体地,所述的有机添加剂在均相溶液中浓度为0.03~0.12mmol/ml,优选为0.03~0.1mmol/ml,进一步优选为0.03~0.06mmol/ml,更进一步优选为0.045mmol/ml。
20.具体地,所述电解质在均相溶液中浓度为0.02~0.08mmol/ml,优选为0.03~0.05mmol/ml,进一步优选为0.03~0.04mmol/ml,更进一步优选为0.03mmol/ml。
21.具体地,所述电催化反应所通入的电流强度为5~20ma;优选地为5~15ma;进一步优选为8ma。
22.具体地,所述电催化反应时间为90~180min,优选为90~160min,进一步优选为120min;反应温度为50~80℃,优选为50~70℃,进一步优选为70℃。
23.进一步地,还包括提纯步骤:反应结束后所得反应液萃取得到有机相,经浓缩,柱层析得到取代吲哚类化合物;
24.萃取为将反应液经乙酸乙酯和饱和氯化钠水溶液萃取;
25.柱层析采用硅胶柱层析;
26.柱层析的洗脱剂为石油醚与乙酸乙酯按照体积比1:0.15的混合溶剂。
27.有益效果:
28.本发明将γ-羟基胺合成取代吲哚和有机电催化技术相结合,使用清洁电能实现氧化还原反应,避免使用氧化或还原试剂。这种电化学吲哚合成策略具有优异的官能团、水和空气耐受性。与现有技术相比,本发明合成方法高效经济,产物中无金属残留、产率高;其在生物医用材料领域应用更有竞争力。
附图说明
29.下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
30.图1为本发明合成取代吲哚化合物的反应方程式。
31.图2是本发明合成取代吲哚化合物的反应装置实物图。
32.图3为实施例1中产物2a的核磁共振氢谱图。
33.图4为实施例1中产物2a的核磁共振碳谱图。
34.图5为实施例2中产物2b的核磁共振氢谱图。
35.图6为实施例2中产物2b的核磁共振碳谱图。
36.图7为实施例3中产物2c的核磁共振氢谱图。
37.图8为实施例3中产物2c的核磁共振碳谱图。
38.图9为实施例4中产物2d的核磁共振氢谱图。
39.图10为实施例4中产物2d的核磁共振碳谱图。
40.图11为实施例5中产物2e的核磁共振氢谱图。
41.图12为实施例5中产物2e的核磁共振碳谱图。
42.图13为实施例6中产物2f的核磁共振氢谱图。
43.图14为实施例6中产物2f的核磁共振碳谱图。
44.图15为实施例7中产物2g的核磁共振氢谱图。
45.图16为实施例7中产物2g的核磁共振碳谱图。
46.图17为实施例8中产物2h的核磁共振氢谱图。
47.图18为实施例8中产物2h的核磁共振碳谱图。
48.图19为实施例9中产物2i的核磁共振氢谱图。
49.图20为实施例9中产物2i的核磁共振碳谱图。
50.图21为实施例10中产物2j的核磁共振氢谱图。
51.图22为实施例10中产物2j的核磁共振碳谱图。
52.图23为实施例11中产物2k的核磁共振氢谱图。
53.图24为实施例11中产物2k的核磁共振碳谱图。
54.图25为实施例11中产物2k的核磁共振氟谱图。
55.图26为实施例12中产物2m的核磁共振氢谱图。
56.图27为实施例12中产物2m的核磁共振碳谱图。
57.图28为实施例12中产物2m的核磁共振氟谱图。
具体实施方式
58.根据下述实施例,可以更好地理解本发明。
59.本发明提供一种由γ-羟基胺电化学合成取代吲哚的方法,将底物1、有机添加剂、电解质和溶剂混合得到的均相溶液,加入到具有阴、阳极的电化学反应装置中,通过电催化一步法合成取代吲哚2。反应方程式如图1所示,反应装置如图2所示。本发明提供如下表1中12个取代吲哚类化合物的具体实施方式。
60.表1
[0061][0062]
实施例1
[0063][0064]
70℃下,将n-(2-(1-羟基-1-苯乙基)苯基)乙磺酰胺(91.5mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。用石油醚与乙酸乙酯10:1的洗脱剂分离,将所得产物真空干燥4h。所得产物2a转化率70%。
[0065]
产物2a的核磁共振氢谱如图3所示,核磁共振碳谱如图4所示:
[0066]
1h nmr(400mhz,dmso-d6)δ7.96-7.90(m,2h),7.84(s,1h),7.76-7.71(m,2h),7.54-7.48(m,2h),7.48-7.43(m,1h),7.42-7.37(m,2h),3.67(q,j=7.3hz,2h),1.11(t,j=7.3hz,3h);13c nmr(101mhz,dmso-d6)δ135.41,132.59,129.02,127.96,127.63,127.44,124.91,124.03,123.62,121.46,120.31,113.33,48.33,7.81;hrms(ei-tof)calcd for c16h15no2s[m]+:285.0800;found:285.0823.
[0067]
实施例2
[0068][0069]
70℃下,将n-(2-(1-羟基-1-苯乙基)苯基)苯磺酰胺(105.9mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2b转化率73%。
[0070]
产物2b的核磁共振氢谱如图5所示,核磁共振碳谱如图6所示:
[0071]
1h nmr(400mhz,dmso-d6)δ8.15-8.14(m,2h),8.12-8.12(m,2h),7.88(d,j=7.9hz,1h),7.78-7.73(m,3h),7.67-7.63(m,2h),7.56-7.53(m,2h),7.49-7.37(m,3h);13c nmr(101mhz,dmso-d6)δ136.91,134.81,134.74,132.29,129.89,129.01,128.47,127.69,127.63,126.88,125.19,124.04,123.63,123.15,120.44,113.52;hrms(ei-tof)calcd for c20h15no2s[m]+:333.0818;found:333.0828.
[0072]
实施例3
[0073][0074]
70℃下,将4-氯-n-(2-(1-羟基-1-苯乙基)苯基)苯磺酰胺(116.1mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2c转化率75%。
[0075]
产物2c的核磁共振氢谱如图7所示,核磁共振碳谱如图8所示:
[0076]
1h nmr(400mhz,dmso-d6)δ8.12-8.07(m,3h),8.03(dt,j=8.3,1.2hz,1h),7.84(dt,j=7.8,1.1hz,1h),7.74-7.70(m,2h),7.69-7.65(m,2h),7.53-7.47(m,2h),7.46-7.32(m,3h);13c nmr(101mhz,dmso-d6)δ139.91,135.61,134.78,132.21,130.11,129.04,128.87,128.58,127.75,125.38,124.26,123.61,123.53,120.55,113.54;hrms(ei-tof)calcd for c20h14clno2s[m]+:367.0428;found:367.0442.
[0077]
实施例4
[0078][0079]
70℃下,将n-(2-(1-羟基-1-苯丙基)苯基)-4-甲基苯磺酰胺(114.3mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2d转化率77%。
[0080]
产物2d的核磁共振氢谱如图9所示,核磁共振碳谱如图10所示:
[0081]
1h nmr(400mhz,dmso-d6)δ8.18(d,j=8.2hz,1h),7.86(d,j=8.4hz,2h),7.55(t,j=7.4hz,2h),7.49-7.35(m,7h),7.28(t,j=8.0hz 1h),2.64(s,3h),2.35(s,3h);13c nmr(101mhz,dmso-d6)δ145.41,135.31,134.99,133.02,132.20,130.38,129.82,129.21,128.81,127.58,126.40,124.49,123.92,121.90,118.96,114.18,21.03,13.40;hrms(ei-tof)calcd for c22h19no2s[m]+:361.1131;found:361.1139.
[0082]
实施例5
[0083][0084]
70℃下,将n-(2-(1-羟基-1,2-二苯基乙基)苯基)-4-甲基苯磺酰胺(132.9mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2e转化率73%。
[0085]
产物2e的核磁共振氢谱如图11所示,核磁共振碳谱如图12所示:
[0086]
1h nmr(400mhz,dmso-d6)δ8.30(d,j=8.4hz,1h),7.54-7.41(m,5h),7.41-7.26(m,10h),7.15(dd,j=7.9,1.6hz,2h),2.34(s,3h);13c nmr(101mhz,dmso-d6)δ145.29,136.58,136.13,134.28,131.89,131.70,130.66,129.97,129.73,129.64,128.67,128.42,127.44,127.31,126.49,125.50,124.59,124.12,119.80,115.55,21.06;hrms(ei-tof)calcd for c27h21no2s[m]+:423.1288;found:423.1304.
[0087]
实施例6
[0088][0089]
70℃下,将n-(2-(2-羟基丁-2-基)苯基)-4-甲基苯磺酰胺(95.7mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2f转化率79%。
[0090]
产物2f的核磁共振氢谱如图13所示,核磁共振碳谱如图14所示。
[0091]1h nmr(400mhz,dmso-d6)δ7.99(d,j=8.2hz,1h),7.60(d,j=8.4hz,2h),7.34(d,j=7.7hz,1h),7.27-7.19(m,3h),7.17-7.13(m,1h),2.43(s,3h),2.17(s,3h),2.01(s,3h);
13
c nmr(101mhz,dmso-d6)δ145.06,135.37,135.07,132.10,130.86,130.19,126.12,124.10,123.45,118.66,116.00,113.99,20.96,12.52,8.53;hrms(ei-tof)calcd for c
17h17
no2s[m]
+
:299.0975;found:299.0977.
[0092]
实施例7
c22h19no3s[m]+:377.1080;found:377.1083.
[0102]
实施例9
[0103][0104]
70℃下,将n-(2-(1-羟基-1-苯乙基)苯基)-[1,1'-联苯]-4-磺酰胺(128.7mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2i转化率73%。
[0105]
产物2i的核磁共振氢谱如图19所示,核磁共振碳谱如图20所示:
[0106]
1h nmr(400mhz,dmso-d6)δ8.21-8.20(m,2h),8.19-8.19(m,1h),8.14(d,j=8.3hz,1h),7.93-7.89(m,3h),7.80-7.78(m,2h),7.73-7.70(m,2h),7.61-7.39(m,8h);13c nmr(101mhz,dmso-d6)δ146.07,137.82,135.55,134.81,132.30,129.10,128.99,128.91,127.99,127.69,127.68,127.62,127.58,127.22,125.23,124.05,123.65,123.14,120.46,113.53;hrms(ei-tof)calcd for c26h19no2s[m]+:409.1131;found:409.1138.
[0107]
实施例10
[0108][0109]
70℃下,将4-溴-n-(2-(1-羟基-1-苯乙基)苯基)苯磺酰胺(129.3mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2j转化率78%。
[0110]
产物2j的核磁共振氢谱如图21所示,核磁共振碳谱如图22所示:
[0111]
1h nmr(400mhz,dmso-d6)δ8.09(s,1h),8.03-7.99(m,3h),7.85-7.81(m,3h),7.74-7.72(m,2h),7.52-7.34(m,5h);13c nmr(101mhz,dmso-d6)δ136.01,134.79,133.08,
132.23,129.12,129.08,128.88,128.60,127.78,125.42,124.30,123.63,123.57,120.59,113.56;hrms(ei-tof)calcd for c20h14brno2s[m]+:410.9923;found:410.9931.
[0112]
实施例11
[0113][0114]
70℃下,将n-(2-(1-羟基-1-苯乙基)苯基)-4-(三氟甲基)苯磺酰胺(126.3mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2k转化率76%。
[0115]
产物2k的核磁共振氢谱如图23所示,核磁共振碳谱如图24所示,核磁共振氟谱如图25所示。
[0116]1h nmr(400mhz,dmso-d6)δ8.33-8.28(m,2h),8.15(s,1h),8.05(dt,j=8.4,1.0hz,1h),7.98(dt,j=8.3,0.9hz,2h),7.84(dt,j=7.9,1.1hz,1h),7.76-7.70(m,2h),7.52-7.46(m,2h),7.46-7.33(m,3h);
13
c nmr(101mhz,dmso-d6)δ140.53,134.75,134.19,133.87,132.07,129.00,128.60,127.97,127.77,127.75,127.14(q,j=3.7hz),125.47,124.37,123.80,123.55,121.67,120.60,113.48;
19
f nmr(376mhz,dmso-d6)δ-61.96;hrms(ei-tof)calcd for c
21h14
f3no2s[m]
+
:401.0692;found:401.0705.
[0117]
实施例12
[0118][0119]
70℃下,将n-(2-(1-羟基-1-苯乙基)苯基)-2-(三氟甲基)苯磺酰胺(126.3mg,0.3mmol,1当量)、对甲苯磺酸(77.5mg,0.45mmol,1.5当量)、四丁基四氟硼酸铵(987.8mg,0.3mmol,1当量)溶解于溶剂acn/tce(6.5+3.5)ml中,得到均相溶液,将上述均相溶液加入配备碳布(40mm x 20mm)阳极和铂片(20mm
×
20mm
×
0.1mm)阴极的10ml三颈圆底烧瓶中,控制电流为8ma,反应时间为120min;反应完成后,产物通过tlc鉴定。真空除去溶剂。提纯步骤同实施例1,所得产物2m转化率72%。
[0120]
产物2m的核磁共振氢谱如图26所示,核磁共振碳谱如图27所示,核磁共振氟谱如图28所示。
[0121]1h nmr(400mhz,dmso-d6)δ8.10(dd,j=7.8,1.4hz,1h),8.05(s,1h),7.95-7.90(m,2h),7.86(td,j=7.9,1.5hz,1h),7.82-7.79(m,1h),7.78-7.74(m,2h),7.70(dd,j=8.0,1.2hz,1h),7.55-7.49(m,2h),7.45-7.36(m,3h);
13
c nmr(101mhz,dmso-d6)δ136.42,135.12,135.07,134.25,132.06,129.84,129.24(d,j=6.1hz),129.04,128.90,128.23,128.16,127.81,127.77,127.68,126.29(d,j=5.9hz),125.99,125.43,124.30,124.03,123.82,122.67,121.10,120.72,113.33;
19
f nmr(376mhz,dmso-d6)δ-55.98;hrms(ei-tof)calcd for c
21h14
f3no2s[m]
+
:401.0692;found:401.0707.
[0122]
对比例1
[0123]
在空气下,将2-烯基苯胺(0.076克,0.20毫摩尔)和碘(iii)试剂(0.140克,0.4毫摩尔)装入聚四氟乙烯密封的反应烧瓶中。向混合物中加入mecn(2.5毫升)。用特氟隆盖密封反应容器。在100℃下剧烈搅拌反应混合物15分钟。通过tlc监测底物的完全消失。在减压下除去溶剂。使用洗脱剂(1∶30乙酸乙酯∶石油醚)通过用二氧化硅干负载纯化粗产物,获得3-(4-甲氧基苯基)-1-甲苯磺酰基-1h-吲哚,转化率为51%。由于使用了金属催化剂,使得金属残留大,在高温下反应,操作危险,且产率低。
[0124]
对比例2
[0125]
将fecl3·
6h2o(5~25mol%)和ddq(0.1~0.2mmol,2当量)添加到压力管中4-甲基-n-[2-[(1e)-2-(4-甲基苯基)乙烯基]苯基]苯磺酰胺(0.05~0.1毫摩尔,1当量)的clch2ch2cl(1~2ml,0.05m)溶液中。在ar气氛下,将所得混合物在80℃下搅拌30分钟。反应完成后,真空浓缩反应混合物。通过硅胶柱色谱纯化残余物(乙酸乙酯∶正己烷=1∶5),得到3-(4-甲基苯基)-1-[(4-甲基苯基)磺酰基]-1h-吲哚,转化率为57%。由于使用了金属催化剂,使得金属残留大,且产率低。
[0126]
本发明提供了一种由γ-羟基胺电化学合成取代吲哚的方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1