用于在癌症中检测腺瘤‑腺癌过渡的方法和组合物与流程
本发明一般性涉及结直肠癌(CRC)和定位肿瘤干细胞病灶及鉴定具有癌症干细胞(CSC)样区和/或具有肿瘤细胞显著扩大的能力的结肠肿瘤的方法。
发明背景
结肠具有多种简单和管状的腺体和分泌粘液的杯状细胞和吸水细胞的混合物。这些腺体称为腺体是由于其将粘液分泌入结肠腔。当这些腺体在基因水平经历多种变化时,其以可预测的方式进展,即从良性进展为侵袭性、恶性结肠癌。
众所周知,遗传变化的逐步取得可表征CRC中肿瘤进展的发展。将腺瘤转化至腺癌的事件的顺序始于调节细胞分裂和细胞凋亡的基因中的多重突变。在一系列的细胞分裂中,突变在细胞中变得越来越普遍,导致异常增生和最终的癌症。这些突变事件与从增生区域至腺瘤区域的离散形态的过渡相关,随后是原位转化和最终的侵袭性癌症。然而该过程每个阶段的遗传驱动因子并未得到完全确认。
结肠组织包含称为隐窝的特定隔室的层,其包含数种不同类型的细胞,包括干细胞。低数目的干细胞存在于这些隐窝的基底处。正常地,这些干细胞产生新的、增殖的细胞,当其转移至隐窝之上并替换较老的细胞凋亡细胞(死细胞)时,其成为分化细胞。正常结肠中上皮的受调节生长由这些定位于隐窝的在休眠和增殖状态间交替变化的肠干细胞(ISC)驱动[1]。Wnt相关的β-连环蛋白(CTNNB1)复合物和通过转化生长因子(TGF)-β和PTEN/PIK3CA/AKT通路的信号传输的亚细胞定位中的移位已显示为影响ISC循环[2,3]。
通过与ISC的类推,已推测癌症干细胞(CSC)的表型和功能上相似的群体在CRC和其他肿瘤中发生[4,5]。然而,如概念性Vogelgram所表示的,CRC中的肿瘤进展更频繁地模式化为:从增生型的粘膜变化至腺瘤区域至原位恶性转化和最终的侵袭性癌症的逐步形态学可观察的过渡[6,7]。遗传变化的逐步获得还表征CRC的发展,且在该模型中,增加的增殖速率和遗传不稳定性导致逐渐更多的异常调节和非生长因子依赖性生长。
至今,CSC模型背后的增殖和休眠的不连续模型如何能容易地与由Vogelgram表示的递增演化模型相协调,这仍然是个问题[8]。此外,Vogelgram过程的每个阶段的遗传驱动因子尚未完全确定且可由于肿瘤异质性而混淆。结直肠腺瘤对比腺癌和癌症通常由评估在肿瘤和周边组织中观察到的多重形态特征的存在或不存在而加以区分。例如,具有腺体良性错位的结直肠腺瘤通常缺少显著的架构和/或细胞非典型且在上皮巢周围持续呈现强IV型胶原蛋白。相比之下,包含侵袭性腺癌的结直肠腺瘤通常呈现显著的架构和/或细胞非典型和弱的、非连续的IV型胶原蛋白。然而,用于区分这些生长类型的方法是多层面的且具有挑战性。
相应地,对容易地评估的结肠肿瘤中关键形态发生过渡的生物标记物进行鉴定是有益的,如在CRC中存在的那些。额外地鉴定发生侵袭性CRC的指征的能力将是理想的。
发明概述
本文提供了用于在结肠肿瘤中鉴定癌症干细胞(CSC)样区的方法,其包括测定结肠肿瘤样品的PTEN表达和检测结肠肿瘤样品中PTEN表达的交替空间模式(alternating spatial pattern)。依据所述方法,PTEN表达的交替空间模式的存在对CSC样区的存在是指示性的。
公开了用于在结肠肿瘤中鉴定腺瘤-腺癌(Ad-ACA)过渡区和用于鉴定包含高度腺瘤和早期腺癌区的结肠肿瘤的方法。这些方法包括测定结肠肿瘤样品的PTEN表达和检测所述结肠肿瘤样品中PTEN表达的交替空间模式。PTEN表达的交替空间模式的存在表明所述肿瘤中的高度腺瘤和早期腺癌区并进一步鉴定所述肿瘤中的腺瘤-腺癌(Ad-ACA)过渡区。
还提供了在结肠肿瘤(如存在于CRC中的)中鉴定CSCS的方法,其包括测定结肠肿瘤样品的PTEN表达和检测所述结肠肿瘤样品中PTEN表达的交替空间模式。在其中检测到PTEN表达的交替空间模式的结肠肿瘤中鉴定CSC,因此表明CSC的存在。在一些实施方案中,将鉴定的CSC进一步从肿瘤组织分离(如从肿瘤组织切片使用例如手动宏观解剖或激光捕获显微切割)。
本发明进一步提供用于诊断具有高度结肠腺瘤和/或早期腺癌的受试者的方法,和用于确定受试者中的结肠肿瘤若不进行治疗将经历侵袭性转化的可能性的方法。这些方法包括测定结肠肿瘤样品的PTEN表达和检测所述结肠肿瘤样品中PTEN表达的交替空间模式。PTEN表达的交替空间模式表明高度结肠腺瘤和早期腺癌的存在且其还表明所述肿瘤若不进行治疗将可能经历侵袭性转化。
在本发明的方法的一些实施方案中,PTEN表达通过在所述结肠肿瘤样品的组织切片上用特异性结合PTEN的抗体实施免疫组化来测定,以检测PTEN蛋白表达。
在一些实施方案中,从具有CRC的受试者中获得结肠肿瘤样品,且所述结肠肿瘤样品是结直肠癌肿瘤样品。
在一些实施方案中,本发明提供的方法进一步包括测定结肠肿瘤的SMAD4表达,和检测结肠肿瘤中SMAD4表达的交替空间模式。测定的SMAD4表达可以是SMAD4蛋白表达且可通过在所述结肠肿瘤样品的组织切片上用特异性结合SMAD4的抗体实施免疫组化来测定。
在一些实施方案中,交替空间的SMAD4表达与交替的PTEN空间表达是负相关的。
在一些实施方案中,由本发明提供的方法进一步包括测定结肠肿瘤的TP53表达,并检测所述结肠肿瘤中TP53表达的交替空间模式;和/或测定结肠肿瘤的CD44表达,并在其中不存在PTEN蛋白表达的区域中检测下调并重新分布至基底上皮边界的CD44的表达;和/或测定结肠肿瘤的ALDH1和EZH2表达,并检测与PTEN蛋白表达平行下调的这些标记物的表达。
在一些实施方案中,本发明的方法进一步包括测定结肠肿瘤的β-连环蛋白表达和检测肿瘤细胞中的β-连环蛋白核转位。核β-连环蛋白表达可与PTEN表达相关。
在一些实施方案中,本发明的方法进一步包括测定结肠肿瘤的下述至少一项:Ki-67表达、MYC表达、MGMT表达和Rel A/p65表达,并检测所述进一步测定的表达中的区域变化。
在一些实施方案中,测定的结肠肿瘤的基因组DNA呈现SMAD4单倍型不足和PTEN单倍型不足的至少一种。
本发明进一步提供用于鉴定具有结直肠癌(CRC)的受试者可能积极响应于用靶向NF-kB通路的疗法的治疗的方法,其包括测定来自受试者的CRC肿瘤样品的PTEN表达和RelA/p65表达,检测所述肿瘤样品中的PTEN表达的交替空间模式和RelA/p65表达的交替空间模式,且当检测到PTEN表达和RelA/p65表达的交替空间模式时将受试者鉴定为可能积极响应于用靶向NF-kB通路的疗法的治疗,由此表明所述受试者可能积极响应于用靶向NF-kB通路的疗法的治疗。
还提供了用于确定具有CRC的受试者会积极响应于用靶向TGF-β通路的疗法的治疗的可能性的方法,其包括测定来自受试者的CRC肿瘤样品的PTEN表达和SMAD4表达,检测肿瘤样品中PTEN表达的交替空间模式和SMAD4表达的交替空间模式,且当检测到所述肿瘤样品中PTEN表达和SMAD4表达的交替空间模式时,将所述受试者鉴定为可能积极响应于用靶向TGF-β通路的疗法的治疗,由此表明所述受试者可能积极响应于用靶向TGF-β通路的疗法的治疗。
本发明的另一方面提供用于鉴定具有18q/SMAD4基因组缺失的个体的方法,其包括测定来自个体的结肠肿瘤样品的SMAD4表达,检测所述肿瘤样品中SMAD4表达的交替空间模式,且当检测到交替空间的SMAD4表达时确定所述个体具有18q/SMAD4基因组缺失。这方面,所述交替的SMAD4空间表达表明肿瘤的基因组DNA可能具有18q/SMAD4基因组缺失。
本发明的另一方面提供用于鉴定具有10q/PTEN基因组缺失的个体的方法,其包括测定来自个体的结肠肿瘤样品,检测所述肿瘤样品中PTEN表达的交替空间模式的存在,且当检测到交替空间PTEN表达时确定所述个体具有10q/PTEN基因缺失。这方面,交替PTEN空间表达表明肿瘤的基因组DNA可能具有10q/PTEN基因组缺失。
在一些实施方案中,测定结肠肿瘤样品的表达包括测定所述肿瘤样品的蛋白表达。测定蛋白表达可包括在所述结肠肿瘤样品的组织切片上用特异性与所述测定的蛋白特异性结合的抗体实施免疫组化。
本发明的另一方面提供试剂盒,其包含至少一种检测组织样品中PTEN表达的试剂。在一些实施方案中,试剂盒进一步包含检测选自下组的至少一种蛋白或核酸的表达的试剂:SMAD4、TP53、ALDH1和CD44。在一个实施方案中,所述试剂为特异性与记载的蛋白结合的抗体且所述试剂盒是包含特异性与PTEN结合的抗体、特异性与SMAD4结合的抗体、特异性与TP53结合的抗体和特异性与CD44结合的抗体的基础试剂盒。
在一些实施方案中,试剂盒进一步包含选自下组的至少一种抗体:特异性与β-连环蛋白结合的抗体、特异性与EZH2结合的抗体、特异性与MYC级俄和的抗体和特异性与RelA/p65结合的抗体。
附图简述
图1.腺瘤(Ad)-腺癌(ACA)过渡时Ki-67和PTEN的交替模式。原发结肠癌切片的Ki-67免疫组化染色(A/上方小图)显示了局限于正常/增生上皮中的隐窝的增殖细胞(左侧)、与侵袭性肿瘤的第一区域相邻的腺瘤区中交替的Ki-67+增殖和Ki-67-休眠细胞的延伸(中部)和侵袭性癌的大部的均匀增殖(右侧)。(B/下方小图)来自相同肿瘤的PTEN免疫染色显示了正常/增生上皮(左侧)、大多腺瘤上皮和侵袭性肿瘤中(右侧)均匀的PTEN表达。Ad-ACA过渡区内的区域显示了开-关交替的PTEN表达(中部)。
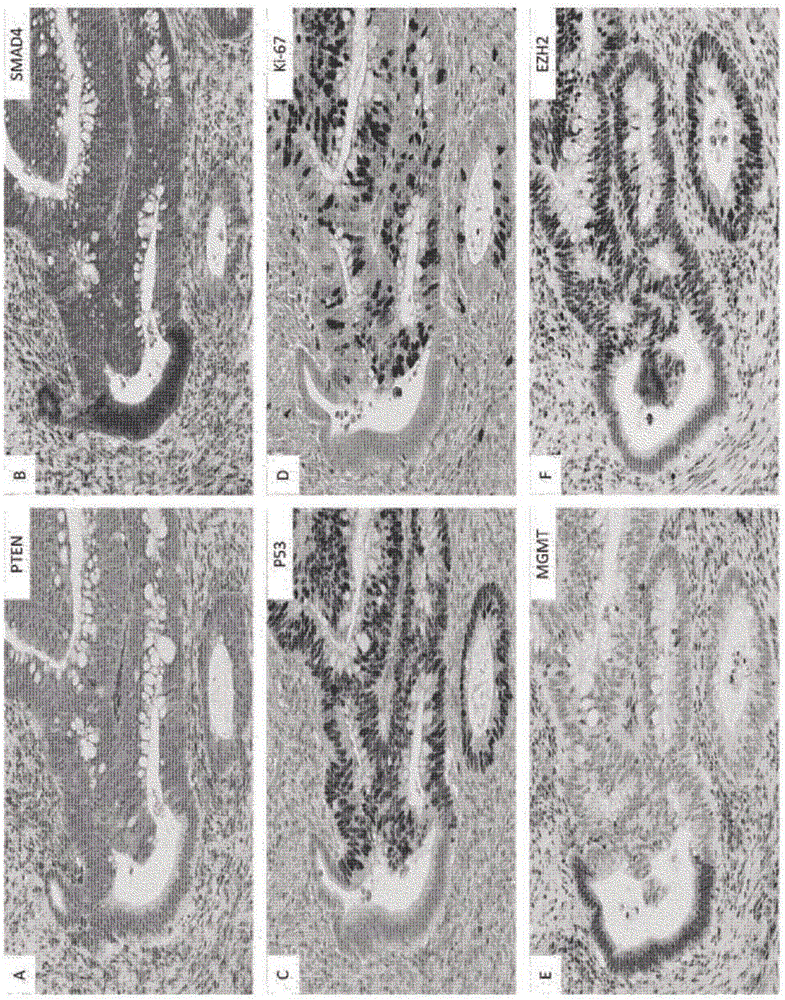
图2.Ad-ACA过渡区中增殖的同步调节和癌症干细胞标记物的灶性(focally)表达。另一原发CRC肿瘤的Ad-ACA过渡区中的免疫染色显示腺体相同部分中PTEN、Ki-67、P53和EZH2的灶性丧失,所述腺体的相同部分显示了SMAD4和MGMT的上调。
图3.具有突出的CSC样Ad-ACA过渡区的肿瘤中多重病灶交替增殖和癌症干细胞标记物表达(A-D)。邻近的腺瘤上皮具有PTEN丢失和减少的Ki-67的突出多重病灶区、具有β-连环蛋白的核定位和CD44的降低水平及基底膜转位。(E,F)另一肿瘤中的Ad-ACA过渡显示了具有EZH2丢失和ALDH1表达的腺瘤上皮的多重病灶延伸。
图4.PTEN缺失与具有突出的CSC样过渡区的肿瘤高度相关。(A)与具有均匀的PTEN下调或正常表达的那些(白色和黄色背景)相比,寡核苷酸/SNP阵列结果显示具有突出的CSC样过渡的CRC中染色体10(黑色虚线箭头)上PTEN基因座的频繁缺失(红线)/通过IHC表示的交替的PTEN丢失(蓝色背景)和完全的PTEN丢失(粉色背景)。涵盖有TP53的染色体17的p臂缺失(中部)和染色体18缺失(右侧)的情况常见于CRC中,无论PTEN IHC的模式如何。(B)CSC样过渡中PTEN IHC的高度放大(上方)以及系列切片的匹配的PTEN FISH图像(下方)显示了上皮细胞中染色体10拷贝数的异质模式与主要的单倍型不足的PTEN丢失一致。
图5.在CSC样过渡区后发生的结肠癌中均匀的TP53异常调节。P53IHC仅显示正常上皮和大多数腺瘤区域中的单细胞/病灶表达,侵袭前/过渡区中的交替表达(小图)和侵袭性腺癌的后期中均匀上调的P53蛋白表达。该情况与图1中所述相同。具有突出的CSC样特征的CRC情形中每种形态学过渡的共同特征示于下文。
发明详述
定义
如本文使用,短语“交替表达”、“交替空间表达”或“表达的交替空间模式”指蛋白表达或核酸表达的多重病灶区域性丢失和获得的空间模式(也描述为空间交替地存在和不存在)。这种交替表达也可以是腺内的。在这方面,“交替的PTEN表达”指空间交替的PTEN蛋白的存在和不存在或交替的PTEN核酸的存在和不存在,如在组织切片中所观察到的(即,和染色的“开/关”重复模式)。
与PTEN表达为“负相关”的“交替的SMAD4表达”指在组织中交替的SMAD4蛋白或核酸表达,其中SMAD4的染色区域或在组织中的存在对应于PTEN不存在的区域,而组织中SMAD4不存在的区域对应于PTEN染色或存在的区域。
如本文使用的“单倍型不足”意为细胞、组织或生物体中仅存在基因的单功能性拷贝,且其他基因拷贝由于突变失活。
如本文使用的“腺瘤”指从腺体产生或类似于腺体的上皮良性肿瘤,且其通常生长成为器官腔。腺瘤可以是带蒂的(具有细长茎的小叶头)或无柄的(广泛基座)。腺瘤息肉是慢性腺瘤的典型。腺瘤增殖特征为不同程度的细胞异常增生(上皮正常分化的异化或丧失)、具有核深染的不规则细胞、(假)分层核、核仁、减少的粘液分泌和有丝分裂。架构可以是管状、绒毛状或管-绒毛状。基底膜和粘膜肌层是完整的。
“腺癌”是具有腺体来源、腺体特征或两者的上皮组织的瘤形成且为腺瘤的恶性对应物。在本发明的一些实施方案中,腺癌是未特别说明的腺癌(腺癌NOS)。在一些实施方案中,结肠腺癌大量表现为团块,其看起来与周边组织颜色不同且肿瘤细胞具有大核并带有突出的核仁。腺癌肿瘤细胞可呈现出有丝分裂发生率的显著增加。
如本文所述的“腺瘤-腺癌交界”(也称为腺瘤-腺癌过渡)指在结肠肿瘤中与侵袭性癌相邻的腺瘤上皮区。
如本文使用的“结肠肿瘤”指结肠中组织的异常肿块,其由细胞的异常生长和分裂导致且包括位于结肠中的良性、恶性前和恶性肿瘤。CRC肿瘤是结肠肿瘤的一种类型。肿瘤细胞的生长超过其周围的正常组织且与周围的正常组织不协调。在一些实施方案中,结肠肿瘤包括结肠上皮细胞。
如本文使用的“癌症干细胞(CSC)样区”或“癌症干细胞(CSC)样腺瘤-腺癌(Ad-ACA)过渡”指Ad-ACA交界区,其在更均匀的侵袭性CRC克隆出现前呈现高度的遗传稳定性和高度的基因组复杂性。该区进一步呈现CSC标记物如β-连环蛋白和/或MGMT的区域变化且可呈现基底重分布的CD44。因此该区的存在表明与恶性转化相关的先导结肠病变的存在和腺瘤-癌过渡的存在。
如本文使用的术语"抗体"或"多种抗体"指全部类型的免疫球蛋白,包括IgG、IgM、IgA、IgD和IgE。抗体可以是单克隆或多克隆的且可以是任何物种来源,包括例如小鼠、大鼠、兔、马、山羊、绵羊或人,或可以是嵌合或人源化的抗体。参见例如Walker等人,Molec.Immunol.26:403-11(1989)。抗体可以是根据美国专利号4,474,893或美国专利号4,816,567中公开的方法产生的重组单克隆抗体。所述抗体还可以根据美国专利号4,676,980公开的方法化学构建。所述抗体还可以是单链抗体或双特异性抗体。
“抗体”包括包含在本发明范围内的抗体片段,其包括例如Fab、F(ab')2和Fc片段和从非IgG的抗体获得的相应片段。这种片段可通过已知的技术产生。例如,F(ab')2片段可通过抗体分子的胃蛋白酶消化产生,且Fab片段可通过减少F(ab')2片段的二硫桥生成。可替换地,可构建Fab表达文库以允许具有合意特异性的单克隆Fab片段的快速和容易的鉴定(Huse等,(1989)Science 254:1275-1281)。
单克隆抗体可在杂交瘤细胞系中根据Kohler和Milstein,(1975)Nature 265:495-97的技术产生。例如,可将包含适当抗原的溶液注射入小鼠且充足的时间后,处死小鼠并获得脾细胞。随后通常在聚乙二醇存在下通过将脾细胞与骨髓瘤细胞或与淋巴瘤细胞融合将脾细胞永生化以产生杂交瘤细胞。杂交瘤细胞随后在适当的介质中生长,并筛选具有合意的特异性的单克隆的上清。单克隆Fab片段可在细菌细胞如大肠杆菌中通过本领域的技术人员已知的重组技术产生。参见例如W.Huse,(1989)Science 246:1275-81。
抗体还可通过本领域的技术人员已知的噬菌体展示技术或通过免疫具有包含感兴趣表位的细胞的异源宿主获得。
如本文使用的术语"特异性结合"或"特异性地结合"指抗体、蛋白或肽与第二化学物质的相互作用时,意为所述相互作用依赖于化学物质上特定结构(例如抗原决定簇或表位)的存在。“特异性结合”或“特异性针对”特定多肽或特定多肽上的表位的抗体是优先与特定多肽或特定多肽上的表位结合的那些(即基本上不与任何其他多肽或多肽表位结合)。例如,抗体识别并特异性结合特定的蛋白结构而非一般性地结合所有蛋白。如果抗体特异性针对表位"A",则在包含标记的"A"和抗体的反应中,含有表位A(或游离的非标记的A)的分子的存在会减少与抗体结合的标记的A的量。
如本文使用的“原位杂交”(ISH)指杂交方法的一种类型,其使用标记的寡核苷酸探针(互补DNA或RNA链)以通过与该靶序列杂交在组织的部分或切片(原位)中定位特异性“靶”DNA或RNA序列。所述探针可在与靶序列杂交前或杂交后直接或间接标记。所述标记是可检测的标记。
“寡SNP基因组阵列”是一类DNA微阵列,其用于检测群体内的多态性如单核苷酸多态性(SNP,其为在DNA中单位点的变化)。寡SNP基因组阵列包括含有固定的核酸靶序列的阵列、一种或多种标记的寡核苷酸探针和记录并解译杂交信号的检测系统。一个示例性的寡-SNP阵列是Genomic Alterations,Postnatal,Oligo-SNP Array(Quest Diagnostics,Madison,New Jersey)。
“分离的”细胞,如分离的CSC,是从其天然存在的组织或生物体中移取的细胞。从组织载玻片或切片分离的CSC已从载玻片或切片分离。尽管分离的细胞可进行纯化,但“分离的”不必需细胞是纯的,而无其他细胞类型或组织组分。
当获得或活检的样品的量不丰富时,组织样品是“有限的”。
“PTEN”指磷酸酶和张力蛋白同源物。PTEN基因在染色体10上位于碱基对89,623,194至碱基对89,728,531。示例性的PTEN氨基酸序列为NCBI/Genbank登录号AAH05821。
“SMAD4”指SMAD家族成员4。SMAD4基因在染色体18上位于碱基对48,556,582至碱基对48,611,411。示例性的SMAD4氨基酸序列为NCBI/Genbank登录号BAB40977。
“TP53”指肿瘤蛋白p53(也称为肿瘤抑制因子p53)。TP53基因在染色体17上位于碱基对7,571,719至碱基对7,590,867。示例性的TP53氨基酸序列为NCBI/Genbank登录号AEX20383。
“CD44”指CD44分子。CD44基因在染色体11上位于碱基对35,160,416至碱基对35,253,948且编码CD44细胞表面糖蛋白。示例性的CD44氨基酸序列是NCBI/Genbank登录号ACI46596。
“Ki-67”指细胞增殖抗原Ki-67。Ki-67示例性的氨基酸序列是Genbank登录号B48666。
“MYC”(也称为c-Myc)指v-myc禽髓细胞瘤病毒致癌基因同源物(homolog)。MYC基因在染色体8上位于碱基对128,748,314至碱基对128,753,679并编码多功能核磷蛋白。示例性的MYC氨基酸序列是NCBI/Genbank登录号AAA59887的氨基酸序列。
“MGMT”指O-6-甲基鸟嘌呤-DNA甲基转移酶。示例性的MGMT氨基酸序列是NCBI/Genbank登录号NP_002403的氨基酸序列。
“Rel A/p65”指v-rel禽网状内皮组织增资病毒致癌基因同源物A(也称为p65或NF-kB p65亚基)。其由在染色体2上位于碱基对61,108,708至碱基对61,171,409的REL基因编码。
“AKT”指v-akt鼠类胸腺瘤病毒致癌基因同源物1(也称为AKT1或PKB)。AKT基因在染色体14上位于碱基对105,235,685至碱基对105,262,079且编码的AKT蛋白为蛋白激酶。
“TCL1”指T-细胞白血病/淋巴瘤蛋白1A。
“EZH2”指zeste同源物2的增强子。其编码EZH2酶,所述酶形成称为多梳抑制复合物-2的蛋白基团的部分。EZH2基因在染色体7上位于碱基对148,504,463至碱基对148,581,440。示例性的EZH2氨基酸序列是NCBI/Genbank登录号AAS09975的氨基酸序列。
“β-连环蛋白”指钙粘蛋白相关蛋白β1,88kDa。其由CTNNB1基因编码,所述基因在染色体3上位于碱基对41,236,400至碱基对41,281,938。
“细胞周期蛋白-D1”也称为CCND1(基因)。CCND1基因编码细胞周期蛋白-D1蛋白,所述蛋白属于高度保守的细胞周期蛋白家族,该家族成员特征是在贯穿细胞周期的蛋白丰度上显著的周期性。CCND1基因在染色体11上位于碱基对69,455,872至碱基对69,469,241。
“ALDH1”指乙醛脱氢酶1家族。
说明
本文描述了用于鉴定结肠肿瘤的方法,所述结肠肿瘤呈现快速空间交替增殖性和低增殖性/休眠的结肠上皮的明显不同的区,其中多种肠干细胞(ISC)/癌症干细胞(CSC)标记物也平行地受到调节。该CSC样过渡区在与侵袭性肿瘤区域相邻的侵袭前腺瘤上皮中发生(即腺瘤-腺癌(Ad-ACA)交界)。这与更深入的、侵袭性腺癌(ACA)和邻近于非肿瘤上皮的腺瘤区域更均匀的增殖和更匀质的表达概貌形成对比。
确定结肠肿瘤是否是与腺癌或侵袭性癌相反的腺瘤对于决定适当的后续采用的操作和/或治疗过程至关重要(即与更激烈的方法如例如化疗相反的简单的息肉移除)。
该CSC样区在显著的百分比的具有形态上完整的Ad-ACA交界的CRC病例中存在。相应地,本文公开的是用于其检测的方法。
不受理论所限,据信其他CRC情况中的该区由于侵袭性组分的过度生长而被除去或由于肿瘤的不完整切片而未取样(且因此而未观察到),或由于一些肿瘤的复杂构架而难于可视化。此外,对于任何给定的标记物,由于遗传突变或基因缺失导致的异常的不存在或过表达可能掩盖该过渡区。这对于其中杂合性(LOH)突变或丢失可导致该蛋白表达的均匀的完全丧失的SMAD4是频繁发生的。因此,本文公开了用于检测具有最高敏感性的Ad-ACA过渡区的标记物组。
在一些情况中,该过渡区显示出通过基因组缺失和TGF-β通路的调节引起的PTEN的单倍型不足,如通过SMAD表达所检测的。该过渡区(其可通过一些肿瘤中的侵袭性组分而过度生长)表现出仅在CRC发展的特定的临时空间阶段中代表肠干细胞(ISC)样表型的激活。
检测方法
相应地,本文提供了用于下述的方法:鉴定结肠肿瘤中CSC样区、鉴定CRC肿瘤中的腺瘤-腺癌(Ad-ACA)过渡区、鉴定包含高度腺瘤和/或早期腺癌区的结肠肿瘤、鉴定结肠肿瘤如CRC肿瘤中的CSC、诊断具有高度结肠腺瘤和/或早期腺癌的受试者,和确定受试者中的结肠肿瘤若不进行治疗将经历侵袭性转化的可能性,所述方法包括测定结肠的PTEN表达和检测所述肿瘤样品中PTEN表达的交替空间模式。
在一些实施方案中,所述方法包括测定结肠或CRC肿瘤样品的PTEN蛋白表达和检测所述肿瘤样品中PTEN蛋白表达的交替空间模式。表达的交替空间模式确立CSC样区在肿瘤组织中Ad-ACA交界处的位置。
空间交替PTEN表达可与癌症干细胞/祖细胞标记物的相似交替空间表达相关,如例如β-连环蛋白/CTNNB1、ALDH1和/或CD44。此外,在一些具有保留的形态过渡的实例中,交替的PTEN表达在位于与侵袭性肿瘤组分紧邻的腺瘤上皮区中观察到,且与交替的Ki-67表达的腺内延伸以及其他细胞循环调节因子和生长调节因子如SMAD4的表达相关。相应地,在一些实施方案中,本发明的方法进一步包括测定至少一、二、三、四种或全部这些蛋白的表达且任选地检测至少一、二、三、四或全部这些蛋白的交替空间表达。
本发明的发明人已鉴定了PTEN-单倍型不足的CRC的不同的CSC样侵袭前过渡阶段,其随后出现具有异常调节的TP53和更稳定的增殖模式及表达概貌的一种或数种侵袭性肿瘤克隆。相应地,本发明的一些实施方案进一步包括测定SMAD4和TP53的至少一种并检测其在肿瘤样品中的交替空间表达。在一些实施方案中,还测定了ALDH1、EZH2、Ki-67、MYC和RelA/p65的一种或多种的蛋白表达且发现其在肿瘤样品中呈现可替换的空间表达。还可测定CD44且可检测下调并重新分布至基底上皮边界(其处于PTEN蛋白表达不存在的区域中)的CD44蛋白表达。
在一些实施方案中,当鉴定Ad-ACA形态学过渡特别是在组织样品有限的情况中,测定一种或多种上述蛋白标记物(如例如PTEN和SMAD4)以补充形态学检查。
本发明的另一方面提供鉴定具有10q/PTEN基因组丢失的个体的方法,其包括测定来自个体的结直肠癌肿瘤样品,检测肿瘤样品中PTEN蛋白表达的交替空间模式,且当检测到交替空间的PTEN蛋白表达时确定个体具有10q/PTEN基因组丢失。
本发明的另一方面提供鉴定具有18q/SMAD4基因组丢失的个体的方法,其包括测定来自个体的结肠或结直肠癌肿瘤样品,检测肿瘤样品中SMAD4蛋白表达的交替空间模式,且当检测到交替空间的SMAD4蛋白表达时确定所述个体具有18q/SMAD4基因组丢失。
本发明的另一方面提供用于鉴定具有CRC的受试者可能积极响应于用靶向TGF-β的疗法或靶向NF-kB通路的疗法的治疗的方法,所述方法通过分别测定结肠肿瘤样品或CRC肿瘤样品中的PTEN和SMAD4或PTEN和p65/RelA并检测其交替空间表达而进行。“积极响应”于治疗意为至少一种CRC可检测的指征在治疗后至少部分减少。
样品
本发明的方法可在结肠组织样品上实施,所述样品如例如结肠肿瘤样品、结肠息肉、CRC肿瘤活检或结肠最内部的内层(粘膜)的另一样品。优选地,测定一个或多个组织样品切片。在一些实施方案中,测定肿瘤的连续切片。
在一些实施方案中,从疑似具有结肠癌的受试者、需要对结肠癌进行诊断或需要对结肠肿瘤分期的受试者和/或正在接受结肠镜的受试者获得肿瘤样品。受试者可以是测试为结肠息肉阳性的和/或可具有结直肠癌。
在具体的实施方案中,从人受试者获得结肠活检并根据本发明的方法分析。当如本文所公开的来检测PTEN和/或其他蛋白的交替空间表达时,可确定需要进一步的活检或探查程序(如例如手术)用于精确诊断或去除未完全切除的息肉的基座或结肠的其他病变。
测定方法
在一些实施方案中,在本文公开的方法中实施的测定包括通过免疫染色来检测组织切片上的蛋白(如例如PTEN)。在一些实施方案中,测定包括在组织切片上用特异性结合感兴趣的蛋白(如例如PTEN)的抗体实施免疫组化(IHC)。抗体可以带有可检测标记或其可与带有可检测标记的化合物结合。可测定以确定其交替空间表达的存在或不存在的蛋白包括一种或多种选自下组的蛋白:ALDH1、b-连环蛋白、CD44、EZH2、c-Myc、细胞周期蛋白-D1、EZH2、MGMT、p53、PTEN、p-SMAD1/5/8和SMAD4。
免疫组化方案为本领域熟知。可用其他方法来检测抗体与蛋白直接或间接的特异性免疫性结合。据此,抗体-对-蛋白的配对体应理解为包括针对待分析蛋白的一抗(例如PTEN或SMAD4)或针对所述一抗的二抗。根据本发明的适当的检测方法的优选实例为冷光,特别是荧光,还有VIS显色和/或放射性发射。
冷光涉及化学发光、生物发光或光致发光导致的光的发射。化学发光涉及化学反应导致的可见光的发射,而生物发光需要荧光素酶的活性。现有的优选光致发光也称为荧光刺激,其由光子吸收引起,优选通过辐射提供,其再次作为具有30-50nm波长跃迁的光子并在约10-8秒的时期内释放。用于荧光检测的仪器包括但不限于常规的台式荧光计、荧光多孔板读取器、光纤荧光计、荧光显微镜和与荧光检测偶联的芯片/微流体系统。
VIS显色指对任何无色物质的可视化,使其对裸眼可见。优选地,显色的强度通过光度计测量。
同位素放射性辐射通过闪烁测量。液体闪烁的过程涉及在样品内对β衰变的检测,所述检测经由称为闪烁鸡尾酒的有机溶剂和溶质系统中的β发射的捕获而进行。样品中的放射性同位素如3H、14C、32P、33P和35S发射的β衰变电子激发溶剂分子,其转而将能量转移至溶质。溶质的能量发射(光子)通过闪烁计数器内的光电倍增管转换为电子信号。鸡尾酒必须还作为保持样品均匀悬浮的增溶剂发挥作用。γ射线光子经常由其他衰变过程(系列衰变)引起,以排除新形成的过剩能量的核。其不具有质量且沿其途径通过碰撞产生极少的直接电离(如果有的话)。吸收γ光子用于检测并通过下述三种机制的一种或多种将其量化:康普顿效应,光电效应和配对生产。本发明有利的γ衰变同位素是125I。
直接的标记物包括附着至抗体的荧光或冷光标签、金属、染料、放射性核素等。可使用由碘-125(125I)标记的抗体。使用特异性针对蛋白的化学发光抗体的化学发光测定适于蛋白水平的敏感、非放射性检测。用荧光团标记的抗体也是适当的。荧光团的实例包括但不限于DAPI、荧光素、Hoechst 33258、R-藻蓝蛋白、B-藻红蛋白、R-藻红蛋白、罗丹明、Texas红和丽丝胺(lissamine)。
间接标记物包括本领域熟知的多种酶,如辣根过氧化物酶(HRP)、碱性磷酸酶(AP)、β-半乳糖苷酶、尿素酶等。抗整合素抗体与酶的共价连接可通过不同的方法实施,如用戊二醛偶联。酶和抗体都与戊二醛经由游离的氨基互相连接,且去除了交织的(networked)酶和抗体的副产物。在另一方法中,如果其为糖蛋白如过氧化物酶,则酶与抗体经由糖残基偶联。酶通过高碘酸钠氧化并直接与抗体的氨基交联。包含糖的其他酶也可以该方式与抗体偶联。酶偶联还可通过将抗体的氨基与酶如β偶半乳糖苷酶的游离巯基使用异双功能接头如琥珀酰亚胺6-(N-马来酰亚氨基)己酸交联实施。可使用辣根过氧化物酶检测系统,例如与发色底物四甲基联苯胺(TMB)一起使用,其在过氧化氢存在下产生在450nm可检测的可溶性产物。碱性磷酸酶检测系统可与例如发色底物磷酸对硝基苯酯一起使用,其产生在405nm可容易检测的可溶性产物。相似地,β-半乳糖苷酶检测系统可与发色底物邻硝基苯基β-D-吡喃半乳糖氧化(ONPG)一起使用,其产生在410nm可检测的可溶性产物。尿素酶检测系统可与底物一起使用,如尿素溴甲酚紫。
用于在组织切片中检测蛋白的抗体可用可检测的部分来标记,其包括但不限于放射性核素、荧光染料例如荧光素、异硫氰酸荧光素(FITC)、OregonGreenTM、罗丹明、Texas红、叔罗丹明异硫氰酸酯(TRITC)、Cy3、Cy5等、荧光标记物(例如绿色荧光蛋白(GFP)、藻红蛋白等)、由肿瘤相关蛋白酶活化的自动淬灭荧光化合物、酶(例如萤光素酶、HRP、AP等)、纳米颗粒、生物素、地高辛等。
在本文提供的方法的测定步骤中一种或多种蛋白标记物可单独或以组合(包括作为双重IHC、三重IHC等)使用。
在一些实施方案中,标记物表达的分析通过荧光原位杂交(FISH)或寡-SNP基因组阵列完成。
在组织切片上检测标记物蛋白或核酸表达的其他方法为本领域熟知且可用于评估在结肠组织中公开的标记物的交替空间表达。
试剂盒
本发明进一步提供用于鉴定结肠组织样品中腺瘤-腺癌过渡区和/或CSC样区(如CRC肿瘤)的试剂盒,其包括检测感兴趣的标记物蛋白的一种或多种试剂。
如本文使用的,“试剂盒”指包装的组分集合,如例如分离的寡核苷酸引物、探针和/或分离的抗体和其他相关试剂。可将试剂盒组分包装于其中的材料的非限制性实例包括盒、袋、信封和管,但试剂盒组分可以其他类型的包装提供给消费者。在一些实施方案中,包含在试剂盒中的其他试剂在试剂盒内的管、小瓶或其他类型的容器中提供。
在一些实施方案中,试剂盒进一步包括用于使用试剂盒组分的说明。所述说明可打印在试剂盒内的材料上或在试剂盒包装上或以电子形式提供。在一些实施方案中,说明描述如何检测表达,例如如何使用包含在试剂盒内的特定组分(例如引物、探针、抗体和/或其他试剂)来检测表达。
在一些实施方案中,试剂盒包含一种或多种检测PTEN蛋白的试剂,和一种或多种检测SMAD4蛋白的试剂。
在一些实施方案中,试剂盒进一步包含一种或多种检测TP53的试剂和/或一种或多种检测CD44的试剂。
在进一步的实施方案中,扩展的试剂盒可额外地包含一种或多种检测至少一种选自下组的标记物的试剂:β连环蛋白、ALDH1、EZH2、MYC和RelA/p65。
在一些实施方案中,试剂特异性检测具体的标记物蛋白。在一些实施方案中,试剂特异性地检测编码具体标记物蛋白的核酸。在一些实施方案中,特异性检测具体蛋白的试剂包括特异性结合该蛋白的抗体。一些试剂盒包含检测一抗的二抗。在一些实施方案中,试剂盒中的抗体包含可检测的标记物或所述抗体直接或间接与含有可检测标记物的化合物结合。
实施例
实施例1-结直肠癌的基因组进展过程中PTEN的下调、检测和再表达
背景/方法:CRC中一般的遗传变化是AKT调节因子PTEN的沉默。然而,发生不同水平的PTEN表明更加复杂的阶段特异性调节。使用免疫组化(IHC)在原发腺癌(ACA)的FFPE切片上研究CRC的PTEN(6H2.1)和TP53(BP53-11),并使用CL PTEN/GRID1探针对PTEN实施FISH。在FFPE提取的DNA上使用CytoScan HD 2.6阵列实施寡SNP阵列(OSA)。
表1
[表1]在结直肠癌中的PTEN发现
结果:在原发CRC中存在4种PTEN表达(表1)。最通常的是在腺瘤区(AD)中完整的PTEN表达和均匀减少的表达(相比非肿瘤上皮),伴随在ACA中逐步衰减的(dimmer)表达。具有PTEN完全丢失的情况不常见且受限于具有最小AD/无AD的ACA。不同的异质(het)组显示PTEN的变化限于ACA中具有PTEN再表达的AD-ACA交界。
通过FISH测得的PTEN基因组缺失在丢失和het组更常见而在完整或下调的情况中是罕见的。
通过OSA,het PTEN的情况大多在遗传上是复杂的,而PTEN基因组缺失(12/18,67%)与18q21/SMAD4(11/18,61%)和17p13/TP53丢失(8/18,44%)相关。当PTEN蛋白在ACA区中再表达4/5(80%)时,在宏观分离的het样品上实施的OSA证明PTEN基因组丢失的逆转;TP53表达也异常调节。
结论:在绝大多数原发CRC中,未观察到PTEN调节或PTEN调节显示在腺瘤阶段下调而无基因组缺失。在该组中,PTEN水平经常随腺瘤-癌过渡而减少,表明逐步的表观遗传沉默。PTEN表达的急剧丢失是不常见的,其与PTEN的缺失相关且处于缺乏Ad-ACA过渡的肿瘤中。在不同的PTEN-het情况中,水平仅在Ad-ACA过渡处波动且再表达随肿瘤进展发生,经常与TP53的异常调节相关。
实施例2-显示干细胞和增殖标记物的高度不连续或快速交替水平的过渡区表征结直肠癌的进展
背景:基因变化的逐步取得表征CRC的进展。这些突变事件与离散的形态学过渡相关,所述形态学过渡是从增生至腺瘤区,随后是原位转化且最终为侵袭性癌。本研究的目的是鉴定CRC中核心形态学过渡的容易评估的生物标记物。
方法:
病例选择和免疫组化:
病例包括提交至Quest Diagnostics Nichols Institute用于分子或免疫分型的原发CRC病例。使用的材料为过量/丢弃的福尔马林固定的石蜡包埋(FFPE)组织切片,其具有研究招入前已匿名的样品,不保留鉴定患者的信息。招入的病例是具有充足FFPE组织切片的原发结肠肿瘤的随机选择。在每个病例中记录从正常至增生粘膜、增生至腺瘤变化(Ad)和腺瘤变化至原位和侵袭性腺癌(ACA)的形态学过渡的位置或不存在。
使用Bond Max III(Leica Microsystems,Wetzlar,Germany),Ultra Benchmark(Ventana,Tucson,AZ)和Link(Dako,Carpinteria,CA)自动化染色平台在4微米FFPE切片上实施免疫组化,其中对于Leica(Epitope Retrieval Solution 2,pH 9.0缓冲剂)和Ventana(Cell Conditioner 1,pH 9.0缓冲剂)在仪器上实施表位检索而对于Dako平台(TRS,pH 9.0缓冲剂持续40分钟)进行离线实施。抗体克隆的列表和工作稀释度列于表2。在应用抗体前对全部载玻片施以过氧化物酶封闭。一抗温育在室温持续12-32分钟,使用3,3-二氨基联苯胺(DAB)检测(DAB,对于Leica包括Bond Polymer Refine,对于Ventana为UltraView DAB且对于Dako为Envision)。载玻片用苏木精复染并用蓝色染剂(bluing)进行后复染。
表2-用于免疫组化的抗体
抗体记载
免疫染色由作者中的两位(KA,DJ)半定量评分,其中使用Aperio XT载玻片扫描仪捕获图像以评估亚细胞染色定位。对于Ki-67,记录具有强核阳性的细胞的类型和数目;PTEN染色评分为:肿瘤中均匀/正常、相比邻近的正常直肠在肿瘤中下调、在肿瘤的全部或部分中的完全/区域性丢失和具有多灶性振荡(其具有与具有正常模式的肿瘤细胞并列的丢失区域)(表3)。对于其他标记物包括ALDH1、β-连环蛋白、CD44、细胞周期蛋白D1、EZH2、MGMT P53、p-SMAD(1/5/8)和SMAD4,针对贯穿肿瘤的水平相对非肿瘤上皮和/或混合淋巴细胞的水平以及亚细胞定位(核、细胞质、膜或组合)评估染色。
表3原发CRC中PTEN免疫组化表达的模式
#p<0.0001,具有保存的Ad-ACA的病例中更低(Fisher精确概率)
*p<0.0001,具有保存的Ad-ACA的病例中更高(Fisher精确概率)
荧光原位杂交:
包含4-μm FFPE切片的载玻片在56℃烘烤过夜,随后脱蜡并使用二甲苯和乙醇脱水。在添加FISH探针前使用0.2N HCl、福尔马林、处理前洗涤和蛋白酶处理载玻片。样品随后在72℃变性5分钟并允许在设置至37℃的湿度箱中杂交过夜(14-18小时)。载玻片使用Metasystems CL PTEN/GRID1,3-色检测探针(MetaSystems,D-5971-100-TC)探测。PTEN基因座特异性探针以橙色标记于染色体条带10q23.3上的315kb区。鸡尾酒中的其他探针包括以绿色标记的针对位于染色体条带10q23.2的GRID1的基因座特异性探针,和以蓝色标记的针对着丝粒控制区(10p11.1-q11.1)的探针。
为建立用于评分的正常截留值,为10个病例计数100个肿瘤细胞,其中计数正常的结肠上皮。如果大于10%的细胞具有单拷贝丧失,则将载玻片分类为杂合缺失,如果大于10%的细胞具有双等位基因丢失则为纯合缺失,且如果少于10%的细胞具有缺失则为正常。通过比较在PTEN/GRID1探针上两个对照区的PTEN信号数评估载玻片(即2R2G2B用于正常、R2G2B用于单倍丢失且2G2B用于双等位基因丢失)。使用上述标准对载玻片计分,联合标记的IHC载玻片以鉴定具有PTEN表达丢失的区。
寡核苷酸/SNP阵列(OSA):
在宏观解剖FFPE肿瘤切片后,使用QiaAmp DNA FFPE组织试剂盒(Qiagen,Valencia,CA)提取基因组DNA,并依照制造商的说明使用CytoScan HD 2.6百万探针微阵列平台(Affymetrix,Santa Clara,CA)评估。用限制酶Nsp I消化至多1μg DNA,与配体连接,并用PCR扩增。随后使用磁性分离技术纯化产物、片段化、并在与微阵列杂交前标记。在PCR纯化步骤评估样品质量。在5个病例中,将具有突出的CSC样Ad-ACA过渡区的样品宏观解剖并独立于更深入的侵袭性ACA进行分析。
使用Chromosome Analysis Suite(ChAS)软件(Affymetrix)分析结果。为比较CSC样病例与具有完整PTEN的病例的IHC模式的频率和通过OSA的异常基因座,使用Fisher精确测试(Fisher’s exact test)生成p值。
突变分析:
在提取自FFPE切片中宏观解剖的肿瘤的基因组DNA上通过基于PCR的DNA Sanger测序实施CTNNB1的外显子3热点和BRAF的外显子11和15的突变分析(具有约10-15%突变等位基因的敏感性),而对于PIK3CA(外显子9和20中的热点)和KRAS(密码子12、13和61)通过焦磷酸测序(具有约2-5%的敏感性)来进行所述突变分析。在具有突出的CSC样过渡区的11个病例中,使用Ampliseq癌症阵列(Life Technologies,Carlsbad,CA)对34个癌症相关基因(其额外包括PTEN、SMAD4和TP53)中的突变热点实施测序。方案为在IonPGM平台上实施的DNA测序的每个制造商的说明且使用SequencePilot软件(JSI MedSystems,Germany)来分析数据。
结果:
增殖和PTEN和SMAD4表达的高度不连续模式表征结直肠肿瘤亚组中的腺瘤-腺癌过渡
检查了原发CR的C良好定向的FFPE肿瘤切片中贯穿形态学过渡的多种增殖标记物的表达模式。在CRC亚组中,在与侵袭性癌(ACA)相邻的区域中鉴定了显示腺瘤上皮(Ad)的Ki-67+增殖性和Ki-67-低增殖性延伸间惊人的腺内改变的局部区。这些不连续的Ki-67表达区经常在10至数百肿瘤细胞的延伸上发生(图1)。尽管IHC模式惊人改变,Ki-67+和Ki-67延伸很大程度在形态上彼此难于区别且在正常/增生上皮过渡或在侵袭性癌中不存在。在检测的亚组中,在这些Ad-ACA过渡区中还观察到了包括细胞周期蛋白D1、MYC和P53的多重细胞周期介导因子的平行不连续表达(图2但未显示)。
生长调节因子PTEN还证明蛋白水平的相似和重叠的剧烈腺内改变,伴随高度离散/不连续的开/关边界(图1B)。这在Ad-ACA交界处的相同区中发生,其中PTEN表达与Ki-67阳性相关(图2)。该通过PTEN IHC测得的开/关Ad-ACA过渡区在研究的全部CRC病例的50/735(6.8%)中是突出的但在具有完整Ad-ACA交界的病例的至少75/202(37.1%)中是灶性存在的(表3)。
该交替模式不同于下调但均匀表达的PTEN IHC模式仍见于大多数CRC中,且在ACA的较小亚组中观察到PTEN的完全丢失模式(表3)。此外,即使在Ad-ACA过渡处具有突出的交替PTEN的病例中,侵袭性ACA总是显示出在侵袭性区域中的PTEN的再表达。这种模式的交替PTEN表达在20个没有相关侵袭性癌的常规结肠腺瘤中未观察到(未显示)。具有通过IHC测得的PTEN表达的急剧且不可逆转的丢失的几乎全部的CRC病例不具有可鉴定的Ad-ACA交界(85/89;95.5%)且还缺乏PTEN表达的上述交替开/关模式。
在检查的病例的较小亚组中,SMAD4,一种TGF-β信号传输调节因子,在这些相同的区中也显示开-关蛋白调节(图2)。连续切片染色和PTEN-SMAD4双重免疫染色(未显示)显示SMAD+和PTEN+细胞间很大程度上可逆的关系。通过在这些区的磷酸化-SMAD1/5/8IHC染色中的核/细胞质转变还提示了TGF-β通路信号传输的动态交替(未显示)。
在Ad-ACA过渡处的多重癌症干细胞标记物和β-连环蛋白/EZH2复合物显示随PTEN的快速交替
鉴于PTEN/AKT信号传输在介导ISC周期循环中的作用,检查了CSC标记物[4,9]在这些过渡区中的水平和亚细胞定位。PTEN-交替增殖/低增殖区也显示干细胞标记物ALDH1、EZH2和MGMT表达、CD44的水平和定位和β-连环蛋白的细胞质/膜的转变及核定位的显著不连续性(图2和3)。在具有低-至-不存在的PTEN和Ki-67阳性的细胞延伸中,β-连环蛋白转移至核(图3A-C)。远离Ad-ACA过渡区,β-连环蛋白在几乎全部的细胞中完全转位至核或存在于膜/细胞质定位中(其中仅疏散的单细胞或小细胞簇具有核定位的蛋白)。在PTEN区中CD44下调且重新分布至基底上皮边界(图3D)。MGMT在显示EZH2和ALDH1染色的下调/不存在的区中上调。
交替标记物表达的准确阶段在肿瘤的Ad-ACA过渡处是可变的。一般而言,Ki-67+增殖区显示PTEN+P53+EZH2低ADLH1-MGMT高SMAD4-的免疫表型(在Ki-67-休眠区中具有相反的表型),如图2中所示。然而,Ad-ACA过渡区中每个标记物的开/关边界的精确重叠是不常见的。更典型地,如图3中所示,为区域关联,其表明对于一些标记物的稍不同的下调或上调阶段。
具有突出的CSC样过渡区的CRC与PTEN的单倍型不足高度相关
在具有突出的CSC样过渡区的CRC病例中,通过OSA检查了整体的基因组发现(表4)并通过对包括PTEN、SMAD4、TP53和CTNNB1的集合进行测序来检查了突变发现。
表4:在原发CRC中通过OSA的基因组发现:具有突出的CSC样过渡的肿瘤与具有完整均匀的PTEN表达的那些的比较
*使用Fisher精确概率(双尾)测试计算p值.使用2x2偶然性表:行为改变的基因座或正常的基因座且列为pTEN IHC丢失/CsC组或下调组.
在具有突出的CSC样扩增的CRC中,如通过PTEN IHC模式所定义的,通过OSA评估的在10q/PTEN的基因组丢失频率(10/16;62.5%)比具有完整/非振动PTEN表达的病例中高得多(6.3%(1/16;6.3%,p=.0007)(图4A)。在CSC样的病例中,当通过FISH评估时,PTEN缺失的频率甚至更高(13/17;76.4%)。使用FISH,还评估了在CSC样区内的个体细胞中的遗传互补。在具有快速交替的PTEN和其他标记物的区中,通过FISH注意到了数个染色体拷贝数的变化以及PTEN拷贝数的变化(图4B/C)。
通过IHC,CSC样肿瘤中PTEN缺失的高频率在这些病例中与显示完全的PTEN丢失的那些相似。然而,在后者的组中,在病例亚组中通过OSA的10q丢失水平与双等位基因缺失一致(未显示)。在全部的组中,可检测的PTEN突变都是不常见的。
在CSC样病例到具有其他PTEN表达模式的病例中,chr 18q/SMAD4 12/16(75%)和chr 17p/TP53 9/16(56.3%)的遗传变化频率相似(图4A)。KRAS突变存在于CSC样病例的83/178(46.7%)中,该频率并不显著区别于其他PTEN表达亚群。在11个具有突出的CSC样过渡区的病例亚组中,TP53(3/11,27.3%)、SMAD4(2/11,18.1%)、APC和CTNNB1突变的频率与未选择的CRC病例中所预期的相似。
表达远离CSC样区的侵袭性CRC中P53的表型性转变;在Ad-ACA交界处和侵袭性肿瘤区室中的表达状态
除了在大多肿瘤中显示更复杂的可变开/关表达模式的MGMT之外,所研究的在Ad-ACA过渡区内交替的标记物在其他肿瘤位置均匀表达。如上文所述,在过渡处具有突出的交替PTEN表达的情况总是在侵袭性肿瘤中再表达蛋白;对于ALDH1和EZH2观察到了相似的模式。然而,SMAD4和CD44表达在侵袭性肿瘤中偶尔完全丢失。然而更惊讶地是P53的表达,在远离Ad-ACA过渡的侵袭性ACA中存在TP53表达的高比率完全丢失(50%)或均匀获得(41.7%)(图5)。在这些肿瘤中,不再观察到CSC样过渡区内所见的交替的P53表达。
检查了CRC的726例中贯穿不同形态学过渡区的生长调节因子和干细胞标记物的增殖和表达模式。在具有保存下来的腺瘤-癌形态学过渡的病例中,鉴定了经常位于紧邻侵袭性组分并延伸其中的腺瘤上皮的特征区,其显示Ki-67+和Ki-67-细胞的快速振荡腺内延伸。该模式与其他细胞周期介导因子和生长调节因子PTEN和SMAD4的振荡表达相关联。腺瘤上皮的这些多重灶性延伸显示交替的阳性/阴性表达边界。其还证明了在多重癌症干细胞(CSC)标记物(包括β-连环蛋白/CTNNB1、MGMT和CD44)的水平和/或亚细胞定位的相似急剧腺内振荡。
相比之下,大多数这些标记物的表达水平在过渡区周围的近端腺瘤和更深入的侵袭性癌中很大程度上是均质性的。该CSC样过渡区,如通过PTENIHC检测,在全部CRC的50/726(6.9%)中是突出的但在具有完整的腺瘤癌交界的97/201(48.2%)病例中至少是灶性存在的。在具有突出的CSC样扩张的CRC上的基因组阵列(ONCOSCANTM HD)和突变分析(AMPLISEQTM)证明了在10/16(62.5%)中的复杂基因组变化,而KRAS、BRAF和CTNNB1突变的频率与未选择的CRC病例中所预期的相似。这些病例中的过渡区还频繁地证明了细胞到细胞的不稳定基因组(如通过FISH所评估),其表明在这些区中的高度遗传不稳定性。
讨论/结论:通过在大量原发CRC肿瘤中检查形态学过渡区,发现了影响多重干细胞和细胞周期的标记物(包括Ki-67、ALDH1、β-连环蛋白、CD44、EZH2、MGMT、MYC、PTEN、P53和SMAD4)的多重灶性交替休眠/增殖腺瘤上皮的局部CSC样过渡区。PTEN-单倍型不足的CRC中的CSC样侵袭前过渡区呈现PTEN/AKT、TGF-β/SMAD和Wnt/β-连环蛋白通路趋同的开-关调节。该瓶颈样区之后是具有完整PTEN表达但具有异常调节的TP53和均匀的高增殖率的侵袭性肿瘤的出现。该过渡区在与侵袭性肿瘤区相邻的侵袭前腺瘤上皮中集中,且与更深入的ACA和与非肿瘤上皮相邻的腺瘤区更均匀的增殖和更均匀的表达概貌形成对比。
尽管如图3中该CSC样区仅在小百分比的CRC中是突出的,其在具有形态学完整的Ad-ACA过渡的情况中以显著的百分比灶性存在,如图2。这些不连续的CSC样区在许多原发CRC中的Ad-ACA交界处的存在表明其检测可作为先兆病变发挥作用,预测初期侵袭性转化。广泛存在的标记物如PTEN和SMAD4可单独或组合使用(包括作为双重IHC)以在对有限的组织样品中的Ad-ACA形态学过渡的鉴定中补充形态学检查。
该区在其他CRC病例中的证据疑似通过侵袭性组分的过度生长而去除或由于不完全的肿瘤切片而未取样,或由于一些肿瘤的复杂结构而难于看到。此外,对于任何给定的标记物,由于基因突变或基因缺失导致的异常不存在或过表达都可掩盖过渡区。其在SMAD4中发生频繁,其中突变或LOH可导致该蛋白表达的均匀完全丢失。因此,需要标记物的集合以检测具有最高敏感性的Ad-ACA过渡区。
多种标记物的开/关边界共定位而非完美重叠表明增殖/休眠过渡的调节可能在这些区相当迅速。在一些病例中看到的这些非同步的边界可能是由于不同的半衰期和每种蛋白的不同下调机制所致,所述下调机制包括蛋白水解性裂解、转录下调和蛋白体清除[10,11]。虽然如此,Ki-67+/PTEN+/MYC+/TP53+区与Ki-67-/MGMT+/β-连环蛋白nuc/SMAD+区交替是在许多病例中可见的特征性模式。
PTEN缺失/单倍型不足与具有突出CSC样区的CRC病例的高度显著相关表明PTEN蛋白的较低水平和导致的作为CSC表型的一个触发因素的AKT激酶活性的差异调节。细胞系和小鼠遗传模型中的多项研究支持了PTEN的单倍型不足或减少的水平在促进癌症进展中的作用[12–14]。此外,由于PTEN的变化或其他因素如TCL1振荡,AKT活性的循环也是癌症相关和非肿瘤干细胞功能及胚胎发生中的共同发现[15–18]。然而,在这些CSC样肿瘤的侵袭性组分中无区域性改变的PTEN朝正常水平的回复表明了在Ad-ACA交界处其他通路的协调性异常调节。
在这方面,在具有总体PTEN IHC丢失的CRC中可检测的Ad/ACA交界的不存在和CSC样交替的表型的不存在表明具有完全的PTEN失活的结肠肿瘤的自然史是不同的[19]。
鉴于在过渡区中观察到的交替的标记物模式,TGF-β/SMAD、PI3K/Akt和Wnt/β-连环蛋白信号传输通路间的激活和抑制性相互作用是有利的[20]。如此处针对PTEN和SMAD4注意到的AKT和BMP/TGF-β信号间的相互作用,在一些模型中是干细胞表型的特征[21–23]。在多个CSC样病例中SMAD信号传输(如通过磷酸化-SMAD抗体所评估的)和SMAD4表达的调节与PTEN相反,可能反映了交界的上皮对BMP/TGF-β信号传输的高度响应性。为帮助驱动过渡区的周期性增殖/休眠特征,SMAD激活对Wnt信号传输的抑制作用[24,25]可抵抗增加的EZH2对驱动β-连环蛋白活性的作用[26]。
在Ad-ACA过渡处的CSC样表型的另一可能的共调节因子是MYC,其也与PTEN、Ki-67和SMAD4平行交替。在细胞系模型中,MYC过表达可通过抑制AKT驱动PTEN上调和EZH2下调[27]。一旦PTEN水平和AKT活性开始振荡,可能发生更广泛的干细胞转录程序效应[28]。PTEN不存在时的AKT活性也已显示出促进遗传不稳定性,其在CSC样过度处观察到且趋于促进遗传进展[29]。
应该注意的是在大多数具有突出的CSC样区的CRC中,如通过均匀的P53IHC丢失或获得所证实的,TP53异常调节在远离过渡区的侵袭性肿瘤中发生。这表明主导的侵袭性CRC克隆的TP53-驱动性衍生物(outgrowth)经常随后发生且可能由CSC样过渡区内的遗传选择所驱动。可能的是这些过渡区是癌症生成的侵袭前阶段的一般现象,其中AKT和Wnt/Beta-连环蛋白周期循环性调节。波动的TP53水平和高度调节的增殖-休眠过渡表明该区可能在更均匀的TP53异常调节侵袭性亚克隆出现前作为肿瘤瓶颈阶段发挥功能。
使用大量的原发性CRC肿瘤,显示出在良好定向的组织切片中,结肠肿瘤显示快速交替增殖和低增殖的结肠上皮的明显不同的区,其中多种ISC/CSC标记物也受到平行调节。该过渡区经常在与具有更均匀的增殖和更均匀的基因和表达概貌的侵袭性肿瘤区相邻的侵袭前腺瘤上皮中开始。该容易评估的表型看起来代表CRC演进中通常发生的遗传不稳定的先兆或过渡阶段。此外,在与肿瘤的侵袭性区紧邻的腺瘤过渡中发生的该区显示出经基因缺失和TGF-β通路的调节的PTEN单倍型不足,如通过SMAD表达所测得的。可能通过一些肿瘤中的侵袭性部分过度生长的该过渡区看起来仅在CRC进展的特定暂时性空间阶段代表ISC样表型的激活。
多重免疫组化标记物包括PTEN、SMAD4、CD44和CTNNB1,强调了该局部CSC样过渡区。
其他实施方案:因此,应该理解的是尽管本发明已由优选的实施方案和其中涵盖的本发明任选的特征、修饰、改进和变化进行了具体公开,本领域的技术人员可采取本公开,且这些修饰、改进和变化应视为在本发明的保护范围内。本文提供的材料、方法和实施例所代表优选的实施方案是示例性的,且并非意在作为对本发明保护范围的限制。
本发明已在本文进行广泛和一般性地描述。落入上位内容的较窄种类和亚群也形成本发明的一部分。其包括具有限制条件或从种类中去除任何对象的负向限制的本发明的一般性描述,无论删去的材料具体记载于本文与否。
此外,当本发明的特征或方面以马库什组描述时,本领域的技术人员将认可本发明也由此就任何单独成员或马库什组成员的亚群进行了描述。
本文提及的全部发表物、专利申请、专利和其他参考文献通过提述以其整体明确地并入,正如其分别通过提述并入的相同程度一样。在冲突的情况中,以本说明书(包括定义)为准。
阐述性地描述于本文的本发明可在不存在任何非本文所具体公开的元素、限制的情况下适当地实践。因此例如,术语"包含"、"包括"、"含有"等将扩大性应当读入而不受限制。此外,本文采用的术语和表述用作说明的术语而非限制的术语,且并非意在使用这些术语和表述来排除任何显示和描述的特征的任何等同或其部分,但应该认可的是在本发明要求保护的范围内的多种修饰是可能的。
参考文献列表
1.Clevers H(2013)Stem Cells:A unifying theory for the crypt.Nature 495:53-54.nature11958[pii];10.1038/nature11958[doi].
2.Pinto D,Clevers H(2005)Wnt control of stem cells and differentiation in the intestinal epithelium.Exp Cell Res 306:357-363.S0014-4827(05)00087-X[pii];10.1016/j.yexcr.2005.02.022[doi].
3.Xia D,Srinivas H,Ahn YH,Sethi G,Sheng X,Yung WK,Xia Q,Chiao PJ,Kim H,Brown PH,Wistuba II,Aggarwal BB,Kurie JM(2007)Mitogen-activated protein kinase kinase-4promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway.J Biol Chem 282:3507-3519.M610141200[pii];10.1074/jbc.M610141200[doi].
4.Hill R,Wu H(2009)PTEN,stem cells,and cancer stem cells.J Biol Chem 284:11755-11759.R800071200[pii];10.1074/jbc.R800071200[doi].
5.Du L,Wang H,He L,Zhang J,Ni B,Wang X,Jin H,Cahuzac N,Mehrpour M,Lu Y,Chen Q(2008)CD44is of functional importance for colorectal cancer stem cells.Clin Cancer Res 14:6751-6760.14/21/6751[pii];10.1158/1078-0432.CCR-08-1034[doi].
6.Cho KR,Vogelstein B(1992)Suppressor gene alterations in the colorectal adenoma-carcinoma sequence.J Cell Biochem Suppl 16G:137-141.
7.Goldberg DM,Diamandis EP(1993)Models of neoplasia and their diagnostic implications:a historical perspective.Clin Chem 39:2360-2374.
8.Hammoud SS,Cairns BR,Jones DA(2013)Epigenetic regulation of colon cancer and intestinal stem cells.Curr Opin Cell Biol 25:177-183.S0955-0674(13)00008-2[pii];10.1016/j.ceb.2013.01.007[doi].
9.Lugli A,Iezzi G,Hostettler I,Muraro MG,Mele V,Tornillo L,Carafa V,Spagnoli G,Terracciano L,Zlobec I(2010)Prognostic impact of the expression of putative cancer stem cell markers CD133,CD166,CD44s,EpCAM,and ALDH1 in colorectal cancer.Br J Cancer 103:382-390.6605762[pii];10.1038/sj.bjc.6605762[doi].
10.Voutsadakis IA(2008)The ubiquitin-proteasome system in colorectal cancer.Biochim Biophys Acta 1782:800-808.S0925-4439(08)00130-0[pii];10.1016/j.bbadis.2008.06.007[doi].
11.Wan M,Cao X,Wu Y,Bai S,Wu L,Shi X,Wang N,Cao X(2002)Jab1 antagonizes TGF-beta signaling by inducing Smad4 degradation.EMBO Rep 3:171-176.10.1093/embo-reports/kvf024[doi];kvf024[pii].
12.Trotman LC,Niki M,Dotan ZA,Koutcher JA,Di CA,Xiao A,Khoo AS,Roy-Burman P,Greenberg NM,Van DT,Cordon-Cardo C,Pandolfi PP(2003)Pten dose dictates cancer progression in the prostate.PLoS Biol 1:E59.10.1371/journal.pbio.0000059[doi].
13.Hill R,Calvopina JH,Kim C,Wang Y,Dawson DW,Donahue TR,Dry S,Wu H(2010)PTEN loss accelerates KrasG12D-induced pancreatic cancer development.Cancer Res 70:7114-7124.0008-5472.CAN-10-1649[pii];10.1158/0008-5472.CAN-10-1649[doi].
14.Byun DS,Ahmed N,Nasser S,Shin J,Al-Obaidi S,Goel S,Corner GA,Wilson AJ,Flanagan DJ,Williams DS,Augenlicht LH,Vincan E,Mariadason JM(2011)Intestinal epithelial-specific PTEN inactivation results in tumor formation.Am J Physiol Gastrointest Liver Physiol 301:G856-G864.ajpgi.00178.2011[pii];10.1152/ajpgi.00178.2011[doi].
15.Narducci MG,Fiorenza MT,Kang SM,Bevilacqua A,Di GM,Remotti D,Picchio MC,Fidanza V,Cooper MD,Croce CM,Mangia F,Russo G(2002)TCL1 participates in early embryonic development and is overexpressed in human seminomas.Proc Natl Acad Sci U S A 99:11712-11717.10.1073/pnas.182412399[doi];182412399[pii].
16.Finkielsztein A,Kelly GM(2009)Altering PI3K-Akt signalling in zebrafish embryos affects PTEN phosphorylation and gastrulation.Biol Cell 101:661-78,4.BC20090034[pii];10.1042/BC20090034[doi].
17.Kharas MG,Okabe R,Ganis JJ,Gozo M,Khandan T,Paktinat M,Gilliland DG,Gritsman K(2010)Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice.Blood 115:1406-1415.blood-2009-06-229443[pii];10.1182/blood-2009-06-229443[doi].
18.Lin Y,Yang Y,Li W,Chen Q,Li J,Pan X,Zhou L,Liu C,Chen C,He J,Cao H,Yao H,Zheng L,Xu X,Xia Z,Ren J,Xiao L,Li L,Shen B,Zhou H,Wang YJ(2012)Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells.Mol Cell 48:627-640.S1097-2765(12)00774-5[pii];10.1016/j.molcel.2012.08.030[doi].
19.Naguib A,Cooke JC,Happerfield L,Kerr L,Gay LJ,Luben RN,Ball RY,Mitrou PN,McTaggart A,Arends MJ(2011)Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study:associations with clinicopathological and dietary factors.BMC Cancer 11:123.1471-2407-11-123[pii];10.1186/1471-2407-11-123[doi].
20.Lee MY,Lim HW,Lee SH,Han HJ(2009)Smad,PI3K/Akt,and Wnt-dependent signaling pathways are involved in BMP-4-induced ESC self-renewal.Stem Cells 27:1858-1868.10.1002/stem.124[doi].
21.Kobielak K,Stokes N,de la Cruz J,Polak L,Fuchs E(2007)Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling.Proc Natl Acad Sci U S A 104:10063-10068.0703004104[pii];10.1073/pnas.0703004104[doi].
22.Tian X,Du H,Fu X,Li K,Li A,Zhang Y(2009)Smad4 restoration leads to a suppression of Wnt/beta-catenin signaling activity and migration capacity in human colon carcinoma cells.Biochem Biophys Res Commun 380:478-483.S0006-291X(09)00133-8[pii];10.1016/j.bbrc.2009.01.124[doi].
23.Jung CJ,Iyengar S,Blahnik KR,Jiang JX,Tahimic C,Torok NJ,de vere White RW,Farnham PJ,Zern M(2012)Human ESC self-renewal promoting microRNAs induce epithelial-mesenchymal transition in hepatocytes by controlling the PTEN and TGFbeta tumor suppressor signaling pathways.Mol Cancer Res 10:979-991.1541-7786.MCR-11-0421[pii];10.1158/1541-7786.MCR-11-0421[doi].
24.He XC,Zhang J,Tong WG,Tawfik O,Ross J,Scoville DH,Tian Q,Zeng X,He X,Wiedemann LM,Mishina Y,Li L(2004)BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling.Nat Genet 36:1117-1121.10.1038/ng1430[doi];ng1430[pii].
25.Freeman TJ,Smith JJ,Chen X,Washington MK,Roland JT,Means AL,Eschrich SA,Yeatman TJ,Deane NG,Beauchamp RD(2012)Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin.Gastroenterology 142:562-571.S0016-5085(11)01622-2[pii];10.1053/j.gastro.2011.11.026[doi].
26.Jung HY,Jun S,Lee M,Kim HC,Wang X,Ji H,McCrea PD,Park JI(2013)PAF and EZH2 Induce Wnt/beta-Catenin Signaling Hyperactivation.Mol Cell 52:193-205.S1097-2765(13)00628-X[pii];10.1016/j.molcel.2013.08.028[doi].
27.Kaur M,Cole MD(2013)MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase.Cancer Res 73:695-705.0008-5472.CAN-12-2522[pii];10.1158/0008-5472.CAN-12-2522[doi].
28.Ning ZQ,Li J,Arceci RJ(2001)Signal transducer and activator of transcription 3 activation is required for Asp(816)mutant c-Kit-mediated cytokine-independent survival and proliferation in human leukemia cells.Blood 97:3559-3567.
29.Mukherjee A,Karmakar P(2013)Attenuation of PTEN perturbs genomic stability via activation of Akt and down-regulation of Rad51 in human embryonic kidney cells.Mol Carcinog 52:611-618.10.1002/mc.21903[doi].
- 还没有人留言评论。精彩留言会获得点赞!