从ΔG的浓度依赖性来测定蛋白质聚集的制作方法
相关申请的交叉引用
本申请要求享有在2014年07月21日提交的美国临时专利申请号62/026,984和在2014年08月07日提交的美国临时专利申请号62/034,504的优先权。上述专利申请的内容以引用的方式全部并入本申请。
背景技术:
蛋白质聚集是影响蛋白质制备,特别是生物学上的长期稳定性的主要问题。通常,聚集体源自在制剂中存在的少量变性或部分变性的蛋白质。这些蛋白质构象倾向于聚集,例如,当蛋白质的疏水核心逐渐暴露于溶剂时。变性蛋白质的聚集逐渐使蛋白质的平衡向增加数量的变性和最终聚集的变性蛋白质移动。在其他情况下,天然状态本身可以显示聚集的倾向,由于连接的小分子药物通常是高度疏水的,这在抗体药物偶联物(adc’s)中是一种加剧的倾向。
因此,存在用于识别和表征蛋白质聚集过程新方法和系统的需求。这样的方法和系统将在(1)鉴定和选择具有最小化聚集和延长长期稳定性的蛋白质制剂和(2)鉴定具有最低聚集倾向的蛋白质变体中提供了极大的益处。本申请解决了这种需求。
技术实现要素:
本发明涉及在尽可能早的时间中确定和表征蛋白质聚集过程的方法和系统以及这些新方法和系统的用途,于(1)鉴定和选择具有最小化聚集和延长长期稳定性的蛋白质制剂,和(2)鉴定具有最低聚集倾向的蛋白质变体。
本发明的第一方面涉及基于所述溶液的蛋白质浓度的溶液中用于蛋白质聚集的量或分数的方法。该方法包括以下步骤:(1)提供包含增加浓度的蛋白质的若干第一溶液;(2)测量若干第一溶液中每个溶液的可观察的特征;(3)基于可观察的属性,确定若干第一溶液中每种浓度下蛋白质的构象稳定性的δg;(4)创建δg与每种浓度的蛋白质之间的相关性;和(5)使用相关性来确定被聚集的蛋白质的量和/或分数和/或被变性的蛋白质的量和/或分数。
如本文所述,“变性”是这样的过程,其中蛋白质通过施加一些外部应力或试剂,例如强酸或碱、浓缩的无机盐、有机溶剂、辐射或热,从而失去存在于其天然状态中的四级结构,三级结构和二级结构。常用的化学变性剂包括尿素和盐酸胍。活细胞中的蛋白质变性可导致细胞活性的破坏和可能的细胞死亡。在体内或体外,变性蛋白质可以表现出宽范围的特征,从构象变化和溶解性的降低到由于疏水基团的暴露导致的聚集。在该过程中,未折叠的蛋白质的暴露的疏水部分可以与其它未折叠的蛋白质的暴露的疏水部分相互作用,自发地导致蛋白聚集。
如本文所述,“蛋白质聚集”是蛋白质在细胞内或细胞外(包括体外)聚集(即,积累和簇集)的现象。当药学上可接受的制剂中的蛋白质聚集时,制剂的有效剂量减少,并且它们可能引起不希望的免疫应答。蛋白质可以在其天然状态或变性状态下聚集。
δg,通过评估蛋白质对增加量的物理或化学变性剂的抗性来测量以确定蛋白质的结构稳定性和/或构象稳定性的热力学参数。物理因子是温度或压力。在这种情况下,通过逐渐增加蛋白质溶液的温度或压力来实现变性。化学因子是变性剂分子,如尿素或盐酸胍。在这种情况下,蛋白质变性通过逐渐增加变性剂的浓度来实现。蛋白质的变性伴随着蛋白质的一些可观察的特征的变化。这些变化的分析用于评估δg。在实施方式中,若干溶液将不含有化学变性剂或化学变性剂的浓度不足以产生可检测水平的变性蛋白质。
在一些实施方式中,可观察的特征是荧光性。当蛋白质变性时,蛋白质的某些可测量的特征也会改变。一个这样的特征是蛋白质的荧光性。荧光性可以是蛋白质固有的,例如通过天然荧光的氨基酸,例如色氨酸,酪氨酸和苯丙氨酸,或者可以于蛋白质是外来的,例如通过包含荧光标记的探针。可以使用其它本领域已知的方法来检测蛋白质的变性状态和/或允许测定δg。除了荧光光谱学之外,有许多物理可观察的特征和其使用的仪器,其对蛋白质的变性程度敏感。这些可观察的特征可以通过各种技术测量,包括但不限于uv/vis光谱、圆二色性、核磁共振(nmr)、红外光谱(ir)和差示扫描量热法等。
在一些实施方式中,所述方法可以进一步包括以下步骤:(1)提供包括增加浓度的蛋白质的若干至少第二溶液;(2)测量若干至少第二溶液中的每种溶液的可观察的特征;(3)基于可观察的特征,确定若干至少第二溶液中每种浓度的蛋白质的构象稳定性的δg;(4)创建于每个若干至少第二溶液的δg与蛋白质的每种浓度之间的相关性;和(5)使用相关性来确定若干至少第二溶液中聚集的蛋白质的量和/或分数和/或变性的蛋白质的量和/或分数。
在实施方式中,所述方法包括将若干第一溶液中每种浓度的蛋白质的构象稳定性的δg与若干至少第二种溶液中每种浓度的蛋白质的构象稳定性的δg进行比较的步骤。
在一些实施方式中,若干至少第二溶液包括若干第二溶液。在一些实施方式中,若干至少第二溶液包括若干第二溶液和若干第三溶液。在一些实施方式中,若干至少第二溶液包括若干第二溶液、若干第三溶液和若干第四溶液。在一些实施方式中,若干至少第二溶液包括若干第二溶液、若干第三溶液、若干第四溶液和若干第五溶液。在一些实施方式中,若干至少第二溶液包括若干第二溶液、若干第三溶液、若干第四溶液、若干第五溶液和更若干溶液(例如,若干第六溶液、若干第七溶液、若干第八溶液、若干第九溶液、若干第十溶液、若干第二十溶液,若干第四十溶液、若干第六十溶液、若干第八十溶液、若干第一百溶液,以及其间的若干任意数)。实施方式包括多于一百个溶液。
若干溶液可以与另一若干溶液在一个或多个(例如,一个、两个、三个、四个、五个、六个、七个、和八个)条件或因素上不同,其可以被调整用于制定溶液。在一些实施方式中,条件或因素选自由缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成、赋形剂浓度、化学变性剂组成和化学变性剂浓度构成的组。在一些实施方式中,若干溶液仅在一个条件或因子上不同于另一若干溶液。在一些实施方式中,若干溶液仅在缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成、赋形剂浓度、化学变性剂组成或化学变性剂浓度不同于另一若干溶液。在一些实施方式中,若干至少第二溶液中的每一种仅在缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成、赋形剂浓度、化学变性剂组成或化学变性剂浓度方面不同于若干第一溶液。在一些实施方式中,每种若干至少第二溶液仅在化学变性剂浓度不同于若干第一溶液。在一些实施方式中,每种若干至少第二溶液包含比若干第一溶液更高浓度的化学变性剂。在一些实施方式中,若干第一溶液不包含化学变性剂或产生不可检测量的或不可检测分数的变性蛋白质的一定量的化学变性剂。
若干溶液的每种溶液可以与若干溶液中的另一溶液在一个或多个(例如,一个、两个、三个、四个、五个、六个、七个、和八个)条件或因子上不同,其可以被调整用于制定溶液(例如,本文所述的条件或因子)。在一些实施方式中,若干溶液中的每种溶液的不同不超过三种条件(例如,无条件、一种条件、两种条件和三种条件),所述条件选自缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成、赋形剂浓度、化学变性剂组成和化学变性剂浓度的组。
在非限制性实施方式中,若干第一溶液中的每种溶液可以具有5.7的ph,包含0.9%nacl和3m尿素,并且若干至少第二溶液中的每种溶液具有6.7的ph,包含0.9%nacl和3m尿素,唯一的区别是若干第一溶液和若干至少第二溶液之间的ph。在另一个非限制性实施方式中,若干第一溶液中的每种溶液可具有5.7的ph,包含1.2%nacl和3m尿素,并且若干至少第二溶液中的每种溶液具有6.7的ph,包含0.9%nacl和4m尿素,其中差异是若干第一溶液和若干至少第二溶液之间的ph和变性剂(即尿素)的浓度。图16显示了可用于本发明的示例性溶液。
如本文所述,“增加浓度”是指本申请的若干溶液(例如,若干第一溶液、若干第二溶液、若干第三溶液等)中的溶液具有一定浓度范围的从最低浓度增加到最高浓度的组分(例如,蛋白质)。例如,若干溶液中的溶液各自地包含0.1μg/ml、0.2μg/ml、0.5μg/ml、1μg/ml、5μg/ml、50μg/ml、100μg/ml、200μg/ml、500μg/ml和1mg/ml浓度的蛋白质。
两个或更多的若干溶液可以包含相同浓度的蛋白质的溶液。在一些实施方式中,若干第一溶液和若干至少第二溶液包含相同浓度的蛋白质的溶液。在一些实施方式中,若干第一溶液和若干第二溶液包含相同浓度的蛋白质的溶液。在非限制性实施方式中,若干第一溶液包括蛋白质的浓度为0.1μg/ml、0.2μg/ml、0.5μg/ml、1μg/ml、5μg/ml、50μg/ml、100μg/ml、200μg/ml、500μg/ml和1mg/ml的溶液,并且若干至少第二溶液包括所述蛋白质的浓度的溶液,其对应于若干第一溶液中的浓度,即各自为0.1μg/ml、0.2μg/ml、0.5μg/ml、1μg/ml、5μg/ml、50μg/ml、100μg/ml、200μg/ml、500μg/ml和1mg/ml。
在一些实施方式中,所述方法可以进一步包括产生蛋白质的构象稳定性的δg和一个或多个(例如,一个、两个、三个、四个、五个、六个、七个和八个)条件或因子之间相关性的步骤,其可以被调整用于制定溶液(例如,本文所述的条件或因素)针对若干第一溶液中的每个溶液以及若干至少第二溶液中的每个溶液。
在实施方式中,所述方法可以进一步包括提供的若干溶液(即“化学变性剂溶液”)具有相同浓度的蛋白质(和/或其他组成成分)和增加浓度的化学变性剂(例如尿素和盐酸胍)。确定化学变性剂的增加浓度对δg,对变性蛋白质的量和/或分数,和/或聚集的蛋白质的量和/或分数的相关性(例如通过测量可观察的特征,其能够监测蛋白质变性)。在非限制性实施方式中,若干溶液包括具有相同浓度的蛋白质和尿素浓度在0和10μm之间,例如0m、1m、2m、3.0m、3.25m、3.5m、3.75m、4m、6m和8m的溶液。
在一些实施方式中,所述方法可以进一步包括比较每种浓度的蛋白质的δg值以确定蛋白质是以其天然状态聚集还是以变性状态聚集的步骤。
在一些实施方式中,所述方法可以进一步包括测定每种不同浓度的变性的蛋白质的量和/或分数的步骤。在一些实施方式中,所述方法可以进一步包括测定每个不同浓度的聚集的蛋白质的量和/或分数的步骤。在一些实施方式中,所述方法可以进一步包括测定变性的蛋白质的量和/或分数和每种不同浓度聚集的蛋白质的量和/或分数的步骤。
在一些实施方式中,所述方法可以进一步包括在若干第一溶液中聚集的蛋白质的量和/或分数,和/或变性的蛋白质的量和/或分数,与在若干至少第二溶液中聚集的蛋白质的量和/或分数,或变性的蛋白质的量和/或分数。
在一些实施方式中,所述方法可以进一步包括产生聚集的蛋白质的量和/或分数和一个或多个(例如,一个、两个、三个、四个、五个、六个、七个和八个)条件或因子之间相关性的步骤,所述条件或因子可以被调整来制定溶液(例如,本文所述的条件或因子)用于若干第一溶液中的每个溶液以及若干至少第二溶液中的每个溶液。在一些实施方式中,所述方法可以进一步包括产生变性的蛋白质的量和/或分数与一个或多个(例如,一个、两个、三个、四个、五个、六个、七个和八个)条件或因子之间相关性的步骤,所述条件或因子可以被调整来制定溶液(例如,本文所述的条件或因子)用于若干第一溶液中的每个溶液以及若干至少第二溶液中的每个溶液。在一些实施方式中,所述方法可以进一步包括产生聚集的蛋白质的量和/或分数与变性的蛋白质的量和/或分数,以及一个或多个(例如一个、两个、三个,四个、五个、六个、七个和八个)条件或因子之间的相关性的步骤,所述条件或因子可以被调整来制定溶液(例如,本文所述的条件或因子)用于若干第一溶液中的每个溶液以及若干至少第二溶液中的每个溶液。
在一些实施方式中,所述方法可以进一步包括使用聚集的蛋白质的量和/或分数与变性的蛋白质的量和/或分数,以及一个或多个(例如一个、两个、三个、四个、五个、六个、七个和八个)条件或因子之间的相关性,其可以被调整来制定溶液(例如,本文所述的条件或因子),来测定用于最小化聚集和/或变性蛋白质的量和/或分数的一个或多个(例如一个、两个、三个、四个、五个、六个、七个和八个))条件或因子。
在一些实施方式中,所述方法可以检测2ng/ml或更少的变性的蛋白质(例如,0.5ng/ml至1.5ng/ml,0.75ng/ml至1.25ng/ml,小于1ng/ml,以及其间的每种浓度)和/或测定2ng/ml或更少的聚集的蛋白质(例如,0.5ng/ml至1.5ng/ml,0.75ng/ml至1.25ng/ml,小于1ng/ml,以及其间的每种浓度)。
δg可以在24小时内测定(例如,小于20小时、小于18小时、小于15小时、小于12小时、小于10小时、小于8小时、小于6小时、小于4小时、小于3小时、小于2小时、小于1小时,以及每次时间之间)。
在一些实施方式中,变性的蛋白质的量和/或分数以及聚集的蛋白质的量和/或分数可以在24小时内测定(例如,小于20小时、小于18小时、小于15小时、小于12小时、小于10小时、小于8小时、小于6小时、小于4小时、小于3小时、小于2小时、小于1小时,以及每次时间之间)。
若干溶液可包括至少3至20种溶液(例如,3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20溶液),每种溶液包含不同浓度的蛋白质。若干溶液中的每种溶液可以包括小于200mg/ml的蛋白质(例如小于100mg、小于50mg/ml、小于20mg/ml、小于10mg/ml、小于5mg/ml、小于2mg/ml、小于1mg/ml、以及其间的任何浓度)。在一些实施方式中,溶液包含0.1μg/ml至小于2mg/ml的蛋白质(例如,小于0.2μg/ml、小于0.5μg/ml、小于1μg/ml、小于2μg/ml、小于3μg/ml、小于4μg/ml、小于5μg/ml、小于10μg/ml、小于20μg/ml、小于50μg/ml、小于100μg/ml、小于200μg/ml、小于500μg/ml、小于1mg/ml,以及其间的任何浓度)。
一种或多种若干溶液可以经受以促进蛋白质变性和/或聚集的条件。在一些实施方式中,变性由化学变性剂(例如,尿素和盐酸胍)诱导。在一些实施方式中,变性通过加热诱导。在一些实施方式中,变性由化学变性剂与热的组合诱导。
在实施方式中,本文所述的方法在体外,间接体内或直接体内进行。在实施方式中,本文所述的方法在体外进行,即在设计的和制备的非天然存在的溶液中进行。在实施方式中,溶液中每种组分/因子和每种组分/因子的量被预先选择并组合以产生溶液。
在一些实施方式中,所述方法可以进一步包括测定蛋白质的浓度的步骤,其中溶液中δg对蛋白质浓度的依赖性最小化。在实施方式中,所述方法还包括鉴定使δg最大化并使δg的蛋白质浓度依赖性最小化的蛋白质浓度的步骤。
在实施方式中,所述方法进一步包括使用δg与若干第一溶液中每种浓度的蛋白质之间的相关性,以确定在所述若干第一溶液中所述变性的蛋白质的量或分数和聚集的所述变性的蛋白质的分数的步骤。在实施方式中,该方法进一步包括使用δg与若干至少第二溶液中每种浓度的蛋白质之间的相关性,以确定在所述若干至少一个溶液中变性的所述蛋白质的量或分数和聚集的所述变性的蛋白质的分数的步骤。实施方式进一步包括比较若干第一溶液中聚集的蛋白质的量或分数或变性的蛋白质的量或分数与若干至少第二溶液中聚集的蛋白质的量或分数或变性的蛋白质的量或分数。
在实施方式中,所述方法还进一步包括选择一种或多种条件进一步最大化δg和最小化δg的蛋白质浓度依赖性的步骤,所述条件选自由缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成和赋形剂浓度构成的组。
上述方面还提供了制备蛋白质制剂(例如,药学上可接受的蛋白质制剂)的方法,所述蛋白质制剂具有在溶液中δg对蛋白质浓度依赖性最小的蛋白质浓度。在实施方式中,制剂具有使蛋白质的构象稳定性的δg最大化并使δg的蛋白质浓度依赖性最小化的蛋白质浓度。在实施方式中,制剂具有蛋白质浓度并且包括使蛋白质的构象稳定性的δg最大化并使δg蛋白质浓度依赖性最小化的一种或多种条件(选自缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成和赋形剂浓度)。在实施方式中,选择蛋白质制剂包括选择使聚集的蛋白质的量或分数最小化和/或使变性的蛋白质的量或分数最小化的一种或多种条件和蛋白质的浓度的步骤。
本发明一个方面是具有如通过本文所述的方法测定的一种或多种条件(选自缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成和赋形剂浓度)和所述浓度的蛋白质(其使蛋白质浓度依赖性δg最小化和/或使聚集的蛋白质的量或分数最小化)的蛋白质制剂(例如药学上可接受的蛋白质制剂)。在实施方式中,制剂包括具有使蛋白质的构象稳定性的δg最大化并使δg的蛋白质浓度依赖性最小化的蛋白质的浓度和一种或多种条件。蛋白质制剂可以包括一种或多种条件和蛋白质的浓度,其使聚集的蛋白质的量或分数最小化和/或使变性的蛋白质的量或分数最小化。
如本文所述,药学上可接受的蛋白质制剂是液体(例如,水性)制备的蛋白质制剂,其对于患者例如动物,优选哺乳动物,更优选人类是稳定的(储存和在体内)和可接受的。药学上可接受的蛋白质制剂是将蛋白质与多种化合物组合以确保储存后稳定的活性药物。这些包括但不限于增溶剂、稳定剂、缓冲剂、张力调节剂、填充剂、粘度增强剂/还原剂、表面活性剂、螯合剂、配体和佐剂。
本发明另一方面涉及用于确定若干蛋白质的变体中稳定的蛋白质的变体方法,其包括以下步骤:(1)提供包括增加浓度的第一蛋白质变体的若干第一溶液;(2)测量若干第一溶液中的每个溶液的可观察的特征;(3)基于可观察的特征,确定若干第一溶液中每个浓度下第一蛋白质变体的构象稳定性的δg;(4)创建δg与第一蛋白质变体的每个浓度之间的相关性;和(5)使用相关性来确定聚集的蛋白质的量或分数(6)提供包括增加浓度的至少第二蛋白质变体的若干至少第二溶液;(7)测量若干至少第二溶液中的每个溶液的可观察的特征;(8)基于可观察的特征,确定若干至少第二溶液中每个浓度下至少第二蛋白质变体的构象稳定性的δg;(9)创建δg与至少第二蛋白质变体的每个浓度之间的相关性,以及(10)使用相关性来确定聚集的蛋白质的量或分数,所述蛋白质变体具有最大化的δg和最小化的δg的蛋白质浓度依赖性和/或具有较少量或部分的聚集的蛋白质被确定为稳定的蛋白质变体。在实施方式中,所述第一溶液中的每种溶液在选自缓冲液组成、缓冲液强度、ph、离子强度、赋形剂组成、赋形剂浓度、化学变性剂和化学变性剂浓度组成的组,并且不同于所述若干至少第二溶液中的每种溶液在一种或多种条件,所述条件选自缓冲液组成,缓冲液强度、ph、离子强度、赋形剂组成、赋形剂浓度、化学变性剂组成和化学变性剂浓度的组。在实施方式中,若干第一溶液中的每种溶液至少在化学变性剂浓度不同,并且若干至少第二溶液中的每种溶液至少在化学变性剂浓度方面不同。
蛋白质变体是在一个或多个氨基酸方面不同,并且这种差异可能影响蛋白质的稳定性(例如,变性和/或聚集的趋势)的蛋白质。蛋白质变体的非限制性实施方式包括从不同杂交瘤克隆获得的单克隆抗体。蛋白质变体也可以是糖基化不同的蛋白质和已经通过小分子的连接(例如共价连接)被修饰的蛋白质;抗体药物偶联物是这种修饰的蛋白质的实例。
如在本说明书和所附权利要求中所使用的,除非上下文另有明确规定,否则单数形式“一”,“一个”和“该”包括复数指示物。
除非特别说明或从上下文显而易见,否则如本文所使用的,术语“或”被理解为包括性的,并且涵盖“或”和“和”两者,除非上下文另有明确指示。
除非特别说明或从上下文显而易见,如本文所使用的,术语“约”理解为在本领域的正常耐受范围内,例如在平均值的2个标准偏差内。大约可以理解为在10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.1%、0.05%表示值。除非另有说明,本文提供的所有数值都用术语“约”修饰。
本文所述的任何方面或实施方式可与本文所公开的任何其它方面或实施方式组合。虽然已经结合其详细的描述说明了本公开,但是前面的描述旨在说明而不是限制本公开的范围,本公开的范围由所附权利要求的范围限定。其他方面,优化和修改在所附权利要求的范围内。
除非另有定义,本文使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的相同的含义。虽然与本文所述的那些类似或等同的其它探针、组合物、方法和试剂盒可用于本发明的实践,但是本文描述了优选的材料和方法。应当理解,本文所使用的术语仅用于描述特定实施方式的目的,而不旨在是限制性的。
本文提及的专利和科学文献建立了本领域技术人员可获得的知识。所有美国专利和本文引用的公开或未公布的美国专利申请通过引用并入本文。本文引用的所有公开的外国专利和专利申请通过引用并入本文。本文引用的所有其它公开的参考文献、文献、手稿和科学文献通过引用并入本文。
从前面的描述中,显而易见的是,可以对本文描述的本发明进行变化和修改以采用它用于各种用途和条件。这些实施方式也在所附权利要求的范围内。
附图说明
专利或申请文件包含至少一张彩色图。具有彩色附图的本专利或专利申请公开的副本将由官方根据请求并支付必要的费用后提供。
图1显示了天然(红色)和变性(绿色)(未折叠或部分未折叠)状态之间的蛋白质的构象平衡。在一些条件下,变性状态(以及天然状态)可能表现出自缔合或聚集的趋势。
图2显示了δg对蛋白质浓度的依赖性,其提供蛋白质在其天然状态或其变性状态下聚集的倾向的定量评估。
图3显示了影响δg的因素。
图4:(a)包括δg和hiv-1蛋白酶浓度之间的关系的图。hiv-1蛋白酶的δg随着蛋白质浓度的增加而增加。(b)包括显示δg和α-胰凝乳蛋白酶浓度之间的关系的图。α-胰凝乳蛋白酶的δg随着蛋白质浓度的增加而降低。
图5包括了显示δg和曲妥珠单抗浓度之间的关系的图。显示了来自两种制剂的数据:曲妥珠单抗在5%葡萄糖溶液(蓝色)和曲妥珠单抗在0.9%nacl溶液(绿色)中。曲妥珠单抗的δg随着5%葡萄糖溶液中蛋白质浓度的增加而增加。
图6包括了显示δg和西妥昔单抗浓度(a)之间,变性西妥昔单抗的分数与西妥昔单抗浓度(b)之间,聚集的西妥昔单抗的分数和西妥昔单抗浓度(c)之间以及聚集的变性西妥昔单抗的分数和西妥昔单抗浓度d)之间关系的图。对于每一组,用于以蓝色显示的数据的溶液与补充有300mml-精氨酸的用红色显示的数据的溶液相同。(a)显示西妥昔单抗的δg随着蛋白质浓度的增加而降低。
图7包括了显示了在两种制剂中测试单克隆抗体的研究的数据的图。该图显示了δg与单克隆抗体浓度(a)之间,变性单克隆抗体部分和单克隆抗体浓度(b)之间,聚集的单克隆抗体部分和单克隆抗体浓度(c)之间,聚集的变性单克隆抗体的部分和单克隆抗体的浓度(d)之间关系的图。(a)显示单克隆抗体的δg随着蛋白质浓度的增加而降低。
图8显示了δg和单克隆抗体-a的蛋白质浓度(左上)之间,变性的单克隆抗体-a的分数和单克隆抗体-a的浓度(右上)之间,聚集的单克隆抗体-a的分数和单克隆抗体-a浓度(左下)之间,对于单克隆抗体-a的ph6.25的制剂的聚集变性单克隆抗体a的分数和单克隆抗体-a的浓度(右下)之间关系的图。
图9显示了δg和单克隆抗体-a的蛋白质浓度(左上)之间,变性的单克隆抗体-a的分数和单克隆抗体-a的浓度(右上)之间,聚集的单克隆抗体-a的分数和单克隆抗体-a浓度(左下)之间,对于单克隆抗体-a的ph6.75的制剂的聚集变性单克隆抗体a的分数和单克隆抗体-a的浓度(右下)之间关系的图。
图10显示了δg和单克隆抗体-a的蛋白质浓度(左上)之间,变性的单克隆抗体-a的分数和单克隆抗体-a的浓度(右上)之间,聚集的单克隆抗体-a的分数和单克隆抗体-a浓度(左下)之间,对于单克隆抗体-a的ph7.75的制剂的聚集变性单克隆抗体a的分数和单克隆抗体-a的浓度(右下)之间关系的图。
图11是8至10的三种制剂的δg的覆盖图。该图显示了单克隆抗体-a的δg随着蛋白质浓度的增加而降低。
图12是8至10的三种制剂的变性的单克隆抗体-a的分数的覆盖图。
图13是8至10的三种制剂的聚集的单克隆抗体-a的分数的覆盖图。
图14是8至10的三种制剂的聚集的,变性的单克隆抗体-a的分数的覆盖图。
图15显示了基于制剂的ph和蛋白质浓度的δg变化的图。理想的蛋白质制剂具有相对于溶剂的最大的δg,相对于时间的恒定的δg,以及相对于蛋白质浓度的恒定的δg。叠加在曲线图上的是具有特定ph的制剂的δg相对于蛋白质浓度图;鉴定在叠加图(左箭头)上的蛋白质浓度是具有最大δg,但是显着的蛋白质浓度依赖性,并且鉴定在叠加图(右箭头)上的蛋白质浓度具有最小的蛋白质浓度依赖性。更后的蛋白质浓度被认为是“多参数最佳条件”。
图16显示了可以包括在本发明中的溶液和若干溶液的非限制性实施方式的图。
具体实施方式
本发明涉及在尽可能早的时间中确定和表征蛋白质聚集过程的方法和系统以及这些新方法和系统的用途,用于(1)鉴定和选择具有最小化聚集和延长长期稳定性的蛋白质制剂,和(2)鉴定具有最低聚集倾向的蛋白质变体。
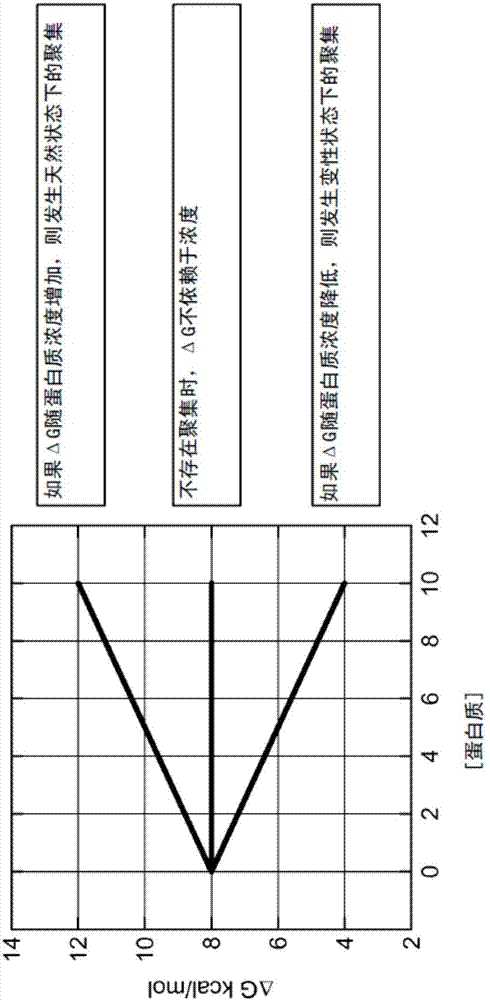
本发明提供了允许快速(例如,在24小时内)定量测定聚集的蛋白质的量的方法。这种新方法基于蛋白质的吉布斯能量稳定性(δg)的浓度依赖性。对在不同蛋白质浓度下测量δg提供了计算在变性或天然状态中聚集的蛋白质的量的必要信息。通常,如果δg随蛋白质浓度降低,则发生变性状态下的聚集,而如果δg随蛋白质浓度增加,则发生天然状态下的聚集。如果δg不随蛋白质浓度变化,则没有聚集的趋势。参见图2。由于δg测量蛋白质的结构稳定性,其后目标是确定最大化δg和同时最小化δg的浓度依赖性的最佳制剂或最佳蛋白质变体。参见图3。本文公开了δg的浓度依赖性的定量分析和在制备蛋白质溶液之后很快估计聚集的方法。早期的结构稳定性和聚集趋势的知识提供了旨在确定最小化蛋白质的聚集和延长蛋白质的长期稳定性的具体措施的关键信息。
使天然状态的蛋白质的结构稳定性最大化并且防止聚集或其它不期望的过程的条件的确定,对于蛋白质治疗剂、蛋白质制剂、过程开发条件、质量控制以及碱性蛋白质的科学发展是必需的。
在蛋白质治疗领域中,存在两个阶段,其中鉴定聚集趋势是关键的:(1)选择用于发展(通常称为发展性评估)的最佳蛋白质变体和(2)确定能确保最长的保质期的制剂条件。生物制剂的长期稳定性通常受到缓慢降低天然蛋白质浓度的变性蛋白质聚集体的出现和缓慢生长的阻碍。这个过程不仅减少了生物活性蛋白质的量,因此降低了治疗效力,而且可以引起不期望的不良反应,包括免疫应答。确定最佳蛋白质变体或最佳制剂需要的能力来测量变性蛋白质的浓度和以及时的方式聚集的蛋白质的分数。使用该信息,可以快速确定具有最佳稳定性和聚集属性的蛋白质变体或制剂。
在这里,考虑两种不同类型的蛋白质:倾向于以其变性状态聚集的蛋白质和倾向于以其天然状态聚集的蛋白质。
i.变性状态聚集
在制剂或蛋白质溶液制备后立即直接测量变性蛋白质的个数是极其困难的。新鲜制剂中变性蛋白质的个数非常小(通常小于总蛋白的0.01%),其通过常规技术下会逃逸检测。尽管这一事实,变性蛋白质的聚集是长期稳定性的主要障碍,因为它缓慢生长并最终破坏蛋白质原料的存活力。常用的评估变性状态聚集的方法,如加速稳定性或强制降解测定,本质上旨在加速变性和聚集群体的积累至可测量水平。这些方法需要几个月,并且不完全可靠,因为用于加速聚集的一些应力不能模仿真实的生活情况。在蛋白质溶液制备之后立即确定变性蛋白质个数的替代方法依赖于天然和变性构象之间的结构平衡的评价。测定吉布斯能量稳定性(δgo)允许计算溶液中变性蛋白质的量非常低的水平(小于总蛋白质的0.001%)。此外,由于聚集和构象平衡是热力学相关联的,我们将展示δgo的蛋白质浓度依赖性提供了评价制剂中蛋白质聚集程度的关键信息。
图1显示了溶液中经历的基本的热力学平衡的蛋白质。天然状态(红色)与变性状态(绿色)平衡存在,其具有以最终不可逆方式聚集的倾向。变性蛋白质(fdenat)的个数或分数等于变性蛋白质(非聚集的和聚集的)的总浓度除以总蛋白质浓度:
其中j是变性聚集体的平均低聚度。
在不存在聚集的情况下,等式1简化为公知的等式:
其在任何恒定温度下仅仅是吉布斯能量稳定性(δgo)的函数。k是也用δgo定义的平衡常数
当蛋白质的聚集或自缔合不存在时,变性的蛋白质的分数与蛋白质浓度无关,因为其仅取决于δgo(等式2的右手侧)。
如果发生聚集,则情况不同,因为聚集是浓度依赖的现象。不受理论束缚,变性蛋白的聚集将增加k的值,而天然的聚集将降低其值;这些变化影响δgo的大小。评价聚集或自缔合的存在或不存在的实验测试是评价不同蛋白质浓度下的δgo。如果δgo与浓度无关,则不存在聚集。如果δgo随浓度降低,则存在变性的蛋白质的聚集,而如果δgo随浓度增加,则存在天然状态的蛋白质的聚集。
用于实验测定δgo的一种方法是差示扫描量热法(dsc)。该方法涉及蛋白质的热变性。由于许多蛋白质,特别是包含绝大多数生物制剂的单克隆抗体的热变性是不可逆过程,因此dsc不能用于测量此类蛋白质的热力学稳定性。因此,dsc的使用限于表现出可逆温度变性的那些蛋白质。
用于实验测定δgo的公认方法包括用化学变性剂例如尿素和盐酸胍测量蛋白质的变性。吉布斯能量已经通过实验显示对化学变性剂浓度是简单线性依赖性的:
δg=δgo-m[变性剂](3)
其中δgo是在零化学变性剂浓度下的蛋白质的稳定性的吉布斯能量;即在没有化学变性剂的实验温度和溶剂条件下蛋白质的稳定性。m是m值。
抗体的化学变性在温度变性不发生的条件下通常是可逆的,因此允许δgo测定。化学变性可以在不同的蛋白质浓度下进行,因此允许直接测量δgo的浓度依赖性。
吉布斯能量的浓度依赖性
如果变性状态具有聚集的倾向(图1),则聚集过程可以根据表观聚集常数
相应地,表观吉布斯能量变为:
在等式5中,
图4b显示了δg和α-胰凝乳蛋白酶浓度的实验浓度依赖性。α-胰凝乳蛋白酶的δg随着蛋白质浓度的增加而降低;因此,α-胰凝乳蛋白酶是在其变性状态下聚集的蛋白质。类似地,在图6a中,显示了δgapp对西妥昔单抗的实验浓度依赖性。在这种情况下,δgapp如在变性状态中聚集的蛋白质所预期的随着蛋白质的浓度降低。
聚集的蛋白质的个数
根据以下标准方程计算聚集的蛋白质的分数:
根据等式5,显然如果其浓度依赖性是已知的,则术语jkj[d]j-1可以仅仅根据测量的δg来表示:
其中
在典型的制剂情况下,绝大多数蛋白质是天然构象,因此变性蛋白质的分数非常小。对于其中δg为8kcal/mol量级的典型蛋白质,变性分数为10-6的量级。从长期稳定性的观点来看,临界量是聚集的变性的蛋白质的分数(fagg的fdenat)。聚集的变性的蛋白质即使最初的量非常小,也将作为在延长的时间段内将使平衡向变性的和聚集的蛋白质的量增加的低点。这种渐进聚集的过程将最终使得溶液无用或在治疗上不可行。聚集的变性的蛋白质的分数由下式给出:
图6b至d显示了可以从图6a中的西妥昔单抗的数据分析中获得的三个部分的个数。类似地,图7a显示δgapp对单克隆抗体的实验浓度依赖性,图7b至d显示了可从图7a中的数据的分析获得的三个部分的个数。此外,图8至图11显示了δgapp对单克隆抗体-a的实验浓度依赖性,图8至图10和图12至图14显示了从实验数据的分析获得的三个部分的个数。
如图所示,变性的蛋白质和聚集的蛋白质的量非常小,如在新鲜制备的溶液中所预期的。变性的部分的量约为10-6。这些低量不可能用常规技术(例如,尺寸排阻色谱和光散射)测量,但可以从δgapp精确计算。从长期稳定性的观点来看,重要的观察的是大多数变性蛋白质聚集体。即使在0.18mg/ml的相对低的浓度下,超过95%的变性的蛋白质也聚集。具有高聚集倾向的变性的蛋白质将最终耗尽天然和活性蛋白质的个数并导致差的长期稳定性。
成功的制剂策略是使变性的蛋白质的量最小化并同时使聚集的变性的蛋白质的比例最小化。从热力学角度看,策略意味着同时最大化δgapp和最小化其蛋白质的浓度依赖性。参见图15
根据上述方法,对于已变性状态聚集的蛋白质,具有相对于溶剂的最大δg,相对于时间的恒定δg,以及相对于蛋白质浓度的δg(即,最小蛋白质浓度依赖性)的理想的蛋白质制剂可以被测定。同样,根据上述方法,对于在变性状态下聚集的蛋白质,具有相对于溶剂的最大δg,相对于时间的恒定δg,以及相对于其它蛋白质的变体的蛋白质浓度的δg(即,最小蛋白质浓度依赖性)的蛋白质变体可以被测定。
ii.天然状态聚集
如果天然状态具有聚集的倾向(图1,左侧),则聚集过程可以根据表观聚集常数kn,i,来描述,类似于上面针对变性状态聚集
相应地,表观吉布斯能量变为:
在等式11中,
天然状态聚集蛋白的个数
根据以下标准方程计算聚集的蛋白质的分数:
如在变性聚集的情况下,如果其浓度依赖性已知,则术语ikn,i[n]i-1可以仅仅根据测量的δg来表示:
其中
在典型的制剂情况下,绝大多数蛋白质是天然构象,因此变性蛋白质的分数非常小。对于其中δg为8kcal/mol量级的典型蛋白质,变性分数为10-6数量级。从长期稳定性的观点来看,临界量是聚集的变性蛋白质的分数(fagg的fdenat)。
图4a显示了δg对hiv-1蛋白酶浓度的实验浓度依赖性;hiv-1蛋白酶的δg随蛋白质浓度的增加而增加;因此,hiv-1蛋白酶是在其天然状态下是二聚体的蛋白质。图5显示了曲妥珠单抗的δgapp的实验浓度依赖性,已知其在5%葡萄糖存在下容易聚集。在这种情况下,δgapp随着蛋白质的浓度增加,如对于以天然状态聚集的蛋白质所预测的。
成功的制剂策略是使变性蛋白质的量最小化并同时使聚集的变性蛋白质的分数最小化。从热力学角度来看,策略意味着同时最大化δgapp和最小化其蛋白质浓度依赖性的策略。
根据上述方法,对于以天然状态聚集的蛋白质,具有相对于溶剂的最大δg,相对于时间的恒定δg,以及相对于蛋白质浓度的δg(即,最小蛋白质浓度依赖性)的理想蛋白质制剂可以被测定。同样,根据上述方法,对于在天然状态下聚集的蛋白质,具有相对于溶剂的最大δg,相对于时间的恒定δg,以及相对于其它蛋白质的变体的蛋白质浓度的δg(即,最小蛋白质浓度依赖性)的蛋白质变体可以被测定。
本发明的系统可以包括具有控制器的装置,该控制器具有处理单元和存储元件。存储元件可以是ram、dram、rom、闪存rom、eerom、磁介质或适于保持计算机可读数据和指令的任何其它介质。指令可以是执行至少本文所述的任何方程的计算所必需的指令。处理单元可以是专用微控制器,个人计算机或任何其他合适的计算设备。此外,该设备具有泵或虹吸系统,其允许其以精确量从多个井中提取液体,并且优选在另一个井中将这些液体混合在一起。该装置还具有测量和记录制剂的荧光的装置,例如通过使用套管将液体吸入市售的荧光检测器中。该装置还包括一个或多个致动器,其可以将套管从一个位置移动到另一个位置,以便从第一井吸取流体并将流体排出到第二井中。这些插管可用于制备产生变性图所需的制剂,并制备蛋白在缓冲液中的制剂。
先前已经描述了本发明中有用的方法,系统和装置,例如在美国专利8,859,295、美国专利8,609,040、美国专利9,029,163和美国专利2012/0045846中,其各自通过引用的方式全部并入本申请。
参考文献
u.b.ericsson,b.m.hallberg,g.t.detitta,n.dekker,p.nordlund,thermofuor-basedhigh-throughputstabilityoptimizationofproteinsforstructuralstudies,anal.biochem,357(2006)289-298.
m.vedadi,f.h.niesen,a.allali-hassani,o.y.fedorov,p.j.finertyjr,g.a.wasney,r.yeung,c.arrowsmith,l.j.ball,h.berglund,r.hui,b.d.marsden,p.nordlund,m.sundstrom,j.weigelt,a.m.edwards,chemicalscreeningmethodstoidentifyligandsthatpromoteproteinstability,proteincrystallizationandstructuredetermination,proc.natl.acad.sci.(usa),103(2006)15835-15840.
m.a.h.capelle,r.gurny,t.arvinte,highthroughputscreeningofproteinformulationstability:practicalconsiderations,eurpeanj.pharmaceuticsandbiopharmaceutics,65(2007)131-148.
g.a.senisterra,p.j.finertyjr.,highthroughputmethodsofassessingproteinstabilityandaggregation,mol.biosyst.,5(2009)217-223.
e.y.chi,s.krishnan,t.w.randolph,j.f.carpenter,physicalstabilityofproteinsinaqueoussolution:mechanismanddrivingforcesinnonnativeproteinaggregation,pharmaceuticalresearch,20(2003)1325-1336.
o.boudker,m.j.todd,e.freire,thestructuralstabilityoftheco-chaperoningroes,journalofmolecularbiology,272(1997)770-779.
m.j.todd,n.semo,e.freire,thestructuralstabilityofthehiv-1protease,journalofmolecularbiology,283(1998)475-488.
r.f.greene,jr.,c.n.pace,ureaandguanidinehydrochloridedenaturationofribonuclease,lysozyme,alpha-chymotrypsin,andbeta-lactoglobulin,j.biol.chem.,249(1974)5388-5393.
c.n.pace,d.v.laurents,r.e.erickson,ureadenaturationofbarnase:phdependenceandcharacterizationoftheunfoldedstate,biochemistry,31(1992)2728-2734.
m.m.santoro,d.w.bolen,unfoldingfreeenergychangesdeterminedbythelinearextrapolationmethod.1.unfoldingofphenylmethanesulfonyl.alpha.chymotrypsinusingdifferentdenaturants,biochemistry,27(1988)8063-8068.
d.w.bolen,m.m.santoro,unfoldingfreeenergychangesdeterminedbythelinearextrapolationmethod.2.incorporationofdgn-uvaluesinathermodynamiccycle,biochemistry,27(1988)8069-8074.
j.k.myers,c.n.pace,j.m.scholtz,denaturantmvaluesandheatcapacitychanges:relationtochangesinaccessiblesurfaceareaofproteinunfolding,prot.science,4(1995)2138-2148.
c.n.pace,determinationandanalysisofureaandguanidinehydrochloridedenaturationcurves,methodsenzymol,131(1986)266-280.
r.m.ionescu,j.vlasak,c.price,m.kirchmeier,contributionofvariabledomainstothestabilityofhumanizedigg1monoclonalantibodies,journalofpharmaceuticalsciences,97(2008)1414-1426.
e.freire,a.schon,b.m.hutchins,r.k.brown,chemicaldenaturationasatoolintheformulationoptimizationofbiologics,drugdiscoverytoday,18(2013)1007-1013.
b.demeule,c.palais,g.machaidze,r.gurny,t.arvinte,newmethodsallowingthedetectionofproteinaggregates:acasestudyontrastuzumab,mabs,1(2009)142-150.
t.arakawa,d.eljima,k.tsumoto,n.obeyama,y.tanaka,y.kita,e.n.timasheff,suppressionofproteininteractionsbyarginine:aproposedmechanismofthearginineeffects,biophyschem,127(2007)1-8.
p.kheddo,m.tracka,j.armer,r.j.dearman,s.uddin,c.f.vanderwalle,a.p.golovanov,theeffectofarginineglutamateonthestabilityofmonoclonalantibodiesinsolution,internationaljournalofpharmaceutics,473(2014)126-133.
m.fukuda,d.kameoka,t.torizawa,m.saitoh,m.yasutake,y.imaeda,a.kioga,a.mizutani,thermodynamicandfluorescenceanalysestodeterminemechanismsofigg1stabilizationanddestabilizationbyarginine,pharmaceuticalresearch,31(2014)992-1001.
a.schon,r.k.brown,b.hutchins,e.freire,ligandbindinganalysisandscreeningbychemicaldenaturationshift,analyticalbiochem,443(2013)52-57.
- 还没有人留言评论。精彩留言会获得点赞!