同时测定锁阳中四种成分的方法与流程
发明领域
本发明属于中药领域,具体涉及中药材质量量化分析技术。
背景技术:
锁阳俗称“不老药”,又名锈铁棒、地毛球、羊锁不拉,为锁阳科植物cynomoriumsongaricumrupr.的干燥肉质茎[1]。分布于甘肃、新疆、内蒙古、青海等地,产于河西走廊沙地、腾格里沙漠、浑善达克沙地西部[2]。具有补肾阳,益精血,润肠通便之功,临床用于肾阳不足,精血亏虚,腰膝痿软,阳痿滑精,肠燥便秘[3]。现代植物化学学者的研究工作表明,锁阳中含有多种类型的活性成分,黄酮类、三萜类、糖类(及糖苷类)、鞣质类、木质素类、有机酸、甾体类以及多种挥发性成分。
锁阳含多种化学成分及药用有效成分,研究中发现锁阳能促进人体新陈代谢,增强免疫调节能力,具有防癌、抗缺氧、抗疲劳、抗衰老、防止心血管疾病,以及抗应激、清除自由基等作用[4-6]。没食子酸、原儿茶酸、儿茶素及根皮苷均为锁阳中的黄酮类化合物,现代药理研究结果显示,锁阳中的没食子酸具有抗炎、抗氧化、抑菌、抗肿瘤及心血管保护作用;原儿茶酸具有增加冠脉流量、降低心肌耗氧量的作用,在体外有明显抑制血小板聚集的活性和抗菌活性;儿茶素和根皮苷具有提高免疫功能、抗氧化、抗病毒、清除自由基、肾脏保护作用[7-10],也与锁阳的主治功能吻合。
2015版药典中仅在锁阳的定性鉴别方面做了规定,而并没有量化分析的方法。近年来,分析测试工作者们在锁阳的化学成分的量化分析方面做了很多工作,研究了其中儿茶素、熊果酸、糖类、鞣质等多类成分的测定方法。王勤等[11]用hplc法测定了锁阳中没食子酸及原儿茶酸的含量,杨瀚春等[12]研究了用hplc法同时测定了锁阳中易溶于水的没食子酸、原儿茶酸、儿茶素的方法,李辰、陈卫林等[13-14]开发出了锁阳中易溶于甲醇的儿茶酸、根皮苷的同时测定法,并建立了醇溶性成分的指纹图谱。但如何实现同时测定溶解度相差较大的多种黄酮类成分,目前暂无学者做过考察。
技术实现要素:
本发明的目的在于为了克服锁阳化学成分测定方面资源及时间的浪费,尤其是同时测定溶解度相差较大的多种黄酮类成分,本发明提供一种新的化学成分测定方法,具体是分次在温水中超声将四种成分同时提出;在高效液相色谱法测定时,流动相采用了较大的梯度范围。通过本发明的方法,可以较完全的将锁阳中的溶解度相差较大的多种黄酮类成分同时提出,并使之呈现在同一张液相色谱图中,避开了几种成分分别测定时的成本消耗和时间消耗。
为实现上述目的,本发明提供的使用hplc法同时测定含锁阳样品(下文也称为供试品)中没食子酸、原儿茶酸、儿茶素及根皮苷四种化学成分的方法,所述方法包括以下步骤:
1)在温水中超声从含锁阳样品中同时提取出没食子酸、原儿茶酸、儿茶素及根皮苷;
2)用高效液相色谱法测定,其中使用流动相为乙腈(a)-0.05%的冰乙酸溶液(b)进行多梯度洗脱;
3)通过与步骤2)所述相同的高效液相色谱法建立没食子酸、原儿茶酸、儿茶素及根皮苷标准品混合物中各自的含量的标准曲线,根据步骤2)得到的高效液相色谱结果,通过上述标准曲线确定所述含锁阳样品中没食子酸、原儿茶酸、儿茶素及根皮苷四种化学成分的含量。
在优选的实施方案中,步骤1)的提取步骤重复数次,优选2-5次,优选3次。
在优选的实施方案中,所述温水温度为40-60℃,优选50-60℃,更优选55℃。在过低的温度,锁阳中各待测成分的提取率不够,温度过高,可能会导致黄酮类成分被高温破坏。
在优选的实施方案中,所述含锁阳样品包括锁阳粉末、含锁阳中成药。
在优选的实施方案中,所述多梯度洗脱包括:
第一梯度:a为5-8%,b为95-92%,
第二梯度:a为8%,b为92%,
第三梯度:a为8-40%,b为92-60%,以及
第四梯度:a为40-5%,b为60-95%。
在优选的实施方案中,所述第一梯度在0~10min中进行,所述第二梯度在10~25min中进行,所述第三梯度在25~80min中进行,所述第四梯度在80~90min中进行。
在优选的实施方案中,所述色谱中使用色谱柱dikmatechnologiesdiamonsilplusc18-a,其中流速为1.0ml/min,检测器为紫外检测器,检测波长为285nm,柱温为30℃,进样体积为5μl。
在优选的实施方案中,所述标准品混合物为1ml含没食子酸100μg、原儿茶酸50μg、儿茶素100μg、根皮苷10μg的混合标准溶液。
在优选的实施方案中,所述方法还任选地包括制备锁阳指纹图谱的步骤。
优选地,所述供试品溶液的制备步骤包括:取粉碎后的锁阳药材粉末(产地甘肃)(过四号筛)约2g,精密称定后,置于50ml的离心管中,加入纯水10ml,超声处理30min(温度控制在55℃左右),摇匀,10000rpm离心10min,移取上清液于25ml的棕色容量瓶中,残渣再依次加水8ml,5ml,在漩涡振荡器上振荡2min后,同上处理两次,合并三次离心的上清液于同一25ml的棕色容量瓶中,加水定容至刻度,摇匀,过滤,取续滤液,即得。
优选地,所述供试品溶液的制备步骤还可以是包括:取含锁阳的中成药肾康宁片20粒,糖衣片除去包衣,精密称定,研细,取约2g,精密称定后,置于50ml的离心管中,加入纯水10ml,超声处理30min(温度控制在55℃左右),摇匀,10000rpm离心10min,移取上清液于25ml的棕色容量瓶中,残渣再依次加水8ml,5ml,在漩涡振荡器上振荡2min后,同上处理两次,合并三次离心的上清液于同一25ml的棕色容量瓶中,加水定容至刻度,摇匀,过滤,取续滤液,即得。
优选地,所述标准品溶液的制备步骤:精密称取没食子酸,原儿茶酸,儿茶素,根皮苷适量于同一容量瓶中,加适量50%的甲醇溶解后,用超纯水稀释制成每1ml含没食子酸100μg、原儿茶酸50μg、儿茶素100μg、根皮苷10μg的混合标准溶液。
优选地,所述乙腈与冰乙酸为色谱纯。
本发明提供的使用hplc法同时测定锁阳中没食子酸、原儿茶酸、儿茶素及根皮苷四种化学成分的方法,也适用于含锁阳中成药锁阳中相关成分的检测。由上述方案可知,本发明所述的hplc法同时测定锁阳中没食子酸、原儿茶酸、儿茶素及根皮苷四种化学成分的方法准确、方法简便、耗时短、重复性好、稳定性高、耐用性强。本发明同时考虑了建立指纹图谱的需要,在保证主要成分提取率的前提下,选用了可以提取尽可能多的成分的供试品制备方法,流动相系统不仅可以保证主要成分与其它成分的分离度达到定量的要求,且其它成分之间也尽可能少重叠,利用本发明的技术,在含量测定的同时可以得到锁阳的指纹图谱,帮助研究者从含量测定除外的方法,分析品种差异及锁阳生长过程中各成分的积累规律。
附图说明
图1纯水空白hplc图谱
图2没食子酸、原儿茶酸、儿茶素、根皮苷混合对照品溶液hplc图谱
图3锁阳供试品溶液hplc图谱
图4没食子酸标准曲线
图5原儿茶酸标准曲线
图6儿茶素标准曲线
图7根皮苷标准曲线
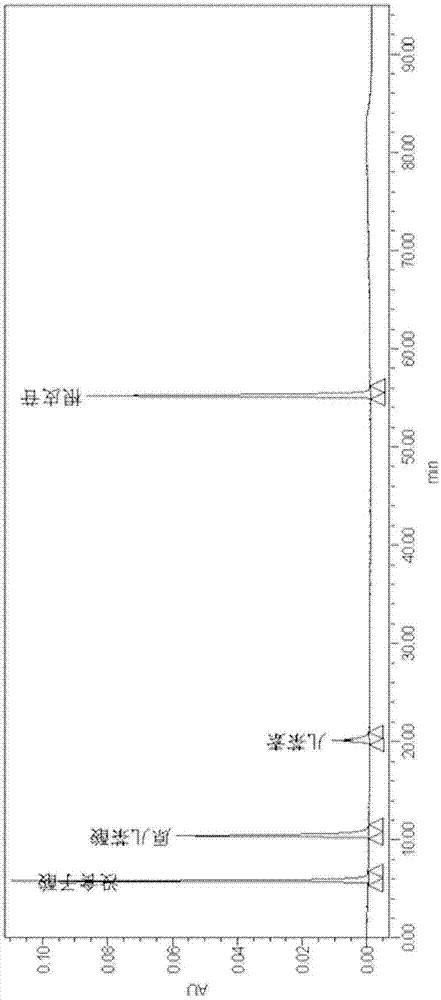
图8肾康宁片供试品溶液hplc图谱
图9锁阳hplc对照指纹图谱,其中3-没食子酸,7-原儿茶酸,14-儿茶素,28-根皮苷
图10锁阳hplc指纹图谱共有模式图
具体实施方式
使用仪器为waterse2695高效液相色谱仪(配有2998pda检测器,empower3色谱工作站);电子分析天平(mettlerab54,mettlerxos205),milli-q超纯水仪(美国millipore公司),超声波提取器(hu-6150b型,天津市恒奥科技发展有限公司)。
儿茶素(110877-200001,购自中国食品药品检定研究院),原儿茶酸(110809-200604,购自中国食品药品检定研究院),没食子酸(110831-200803,纯度90.1%,购自中国食品药品检定研究院),根皮苷(y-029-150705,纯度≥98%,购自成都瑞芬思生物科技有限公司),乙腈(色谱纯,merck公司),冰乙酸(色谱纯,天津康科德科技有限公司)。
色谱条件:色谱柱dikmatechnologiesdiamonsilplusc18-a(5μm,4.6mm×250mm);以乙腈为流动相a,以0.05%冰乙酸溶液为流动相b,分多个梯度进行洗,其中第一梯度(0~10min)中a为5-8%,b为95-92%,第二梯度(10~25min)中a为8%,b为92%,第三梯度(25~80min)中a为8-40%,b为92-60%,第四梯度(80~90min)中a为40-5%,b为60-95%;流速1.0ml/min,检测波长285nm,柱温为30℃,进样量5μl,理论塔板数按没食子酸计算应不低于4000。按外标法以峰面积计算含量。
供试品溶液的制备:取粉碎后的锁阳药材粉末(产地甘肃)(过四号筛)约2g,精密称定后,置于50ml的离心管中,加入纯水10ml,超声处理30min(温度控制在55℃左右),摇匀,10000rpm离心10min,移取上清液于25ml的棕色容量瓶中,残渣再依次加水8ml,5ml,在漩涡振荡器上振荡2min后,同上处理两次,合并三次离心的上清液于同一25ml的棕色容量瓶中,加水定容至刻度,摇匀,过滤,取续滤液,即得。
对照品溶液的制备;精密称取没食子酸,原儿茶酸,儿茶素,根皮苷适量于同一容量瓶中,加适量50%的甲醇溶解后,用超纯水稀释制成每1ml含没食子酸100μg、原儿茶酸50μg、儿茶素100μg、根皮苷10μg的混合标准溶液。
实施例1锁阳药材含量测定
空白试验:考虑到本实验的供试品处理过程,将超纯水作为空白样品即可。空白样品色谱图如附图1所示,比较图1、图2可见,在测定范围内,空白样品不影响供试品的测定,锁阳供试品溶液hplc图谱如图3所示。
标准曲线的制备:精密称取没食子酸(110-831-201605,90.8%,中国食品药品检定研究院)10.97mg,原儿茶酸(110.809-200604,中国食品药品检定研究院)4.75mg,儿茶素(877-200001,中国食品药品检定研究院)10.52mg,根皮苷(y-029-150705,98.96%,成都瑞芬思生物科技有限公司)7.16mg于25ml的棕色容量瓶中,加入50%的甲醇适量使溶解,并以50%甲醇定容至刻度,得混合标准母液,用超纯水将此混合标准母液梯度稀释,得表1中的系列标准混合工作液。将系列标准混合工作液进高效液相色谱仪测定,进样体积5μl,以标准工作液浓度c为横坐标,各峰面积a为纵坐标,绘制标准曲线(对于没食子酸、原儿茶酸、儿茶素和根皮苷,分别由图4-7表示)。表2~表5列出了各成分的直线范围及标准曲线方程。
表1锁阳系列标准混合溶液表
表2没食子酸标准曲线数据
表3原儿茶酸标准曲线数据
表4儿茶素标准曲线数据
表5根皮苷标准曲线数据
仪器稳定性:精密称取没食子酸(110831-200803,90.1%,中国食品药品检定研究院)6.72mg,原儿茶酸(110809-200604,中国食品药品检定研究院),儿茶素(877-200001,中国食品药品检定研究院)5.36mg,根皮苷(y-029-150705,98.96%,成都瑞芬思生物科技有限公司)6.78mg于同一25ml的容量瓶中,得到混合标准溶液母液,并将此母液稀释2.5倍,得到混合标准工作液,将此工作液连续6次进高效液相色谱仪测定其中各峰面积,计算6次测得峰面积的rsd值,如表6所示,各成分的rsd值均小于3%,表明仪器处于稳定运行状态。
表6仪器稳定性测试结果
方法重复性:称取6号锁阳样品(产自甘肃省肃南县)6份,按照供试品制备方法,平行制备6份供试品溶液,按照之前所述测定法,测定其中各成分的含量值,计算6次测定结果的rsd,如表7所示,四种成分测定结果的rsd值均符合中国药典2015年版四部9101(药品质量标准分析方法验证指导原则)关于方法重复性的要求。
表7重复性测定数据
中间精密度:由本实验三位分析人员按照试验确定的方法对6号锁阳样品(产自甘肃省肃南县)中的各成分进行测定,计算结果的rsd值,由表8中的测定数据可见,本发明的测定方法在不同实验人员操作时,稳定性良好。
表8中间精密度
方法准确度:称取6号锁阳样品(产自甘肃省肃南县)约1g,平行称取6份,精密称定后,在每份样品中各加入一定量的标准溶液,用纯水补足10ml,按照供试品溶液制备方法制备加标溶液,测定其中各成分的含量,计算各成分的加标回收率。各成分的加标回收率均符合中国药典2015年版四部9101(药品质量标准分析方法验证指导原则)的规定,测定结果见表9~表12。
表9锁阳中没食子酸的加标回收数据
表10锁阳中原儿茶酸的加标回收数据
表11锁阳中儿茶素的加标回收数据
表12锁阳中根皮苷的加标回收数据
样品稳定性:称取6号锁阳样品(产自甘肃省肃南县)约2g,精密称定后,按照供试品溶液制备方法制备供试品溶液,将此供试品溶液在0h、2h、4h、6h、8h、10h、12h、16h、20h、24h、30h、36h、42h、48h进液相色谱仪测定,记录各次测定的没食子酸、原儿茶酸、儿茶素和根皮苷的峰面积,计算供试品溶液的稳定性,四种成分的峰面积在48h内的rsd%值均小于3%,表明供试品溶液在48h内稳定。测定结果见表13。
表13样品稳定性数据
检出限和定量限:根据s/n=3及s/n=10时的标样浓度,计算得到称样量为2g,hplc测定进样体积为5μl时,没食子酸、原儿茶酸、儿茶素和根皮苷的检出限和定量限。结果见表14,实际测定时,可以根据供试品中各成分的含量高低,适当调整称样量及供试品溶液进样量。
表14检出限及定量限数据
样品测定:取18批锁阳样品,按照前述方法分别制备供试品溶液,在前述色谱条件下测定,精密吸取混合对照品溶液和供试品溶液,注入高效液相色谱仪,记录各次进样中四种成分的峰面积,外标法定量,结果见表15。
表15锁阳样品中的四种成分的含量
实施例2中成药中多种成分的测定
本实施例与实施例1基本相同,区别在于用中成药肾康宁片(批号:1601002)代替锁阳药材,供试品溶液制备方法稍有改变,肾康宁供试品溶液色谱图如图8所示。(图不是结果,测定结果是数据)
供试品溶液的制备:取含锁阳的中成药肾康宁片20粒,糖衣片除去包衣,精密称定,研细,取约2g,精密称定后,置于50ml的离心管中,加入纯水10ml,超声处理30min(温度控制在55℃左右),摇匀,10000rpm离心10min,移取上清液于25ml的棕色容量瓶中,残渣再依次加水8ml,5ml,在漩涡振荡器上振荡2min后,同上处理两次,合并三次离心的上清液于同一25ml的棕色容量瓶中,加水定容至刻度,摇匀,过滤,取续滤液,即得。
方法重复性:称取研细后的肾康宁片样品6份,按照供试品制备方法,平行制备6份供试品溶液,测定其中各成分的含量值,计算6次测定结果的rsd。测定结果见表16,各成分重复测定的rsd值符合中国药典2015年版四部9101(药品质量标准分析方法验证指导原则),表明本专利发明的测定方法稳定性良好。
表16重复性测定数据
方法准确度:称取研细后的肾康宁片粉末约1g,平行称取6份,精密称定后,在每份样品中各加入一定量的标准溶液,用水补足10ml,按照供试品溶液制备方法制备加标溶液,测定其中各成分的含量,计算各成分的加标回收率。测定结果见表17~表20。各成分的回收率符合中国药典2015年版四部9101(药品质量标准分析方法验证指导原则)的要求,表明本专利发明的测定方法用于测定中成药肾康宁片的准确度较高。
表17肾康宁片中没食子酸的加标回收数据
表18肾康宁片中原儿茶酸的加标回收数据
表19肾康宁片中儿茶素的加标回收数据
表20肾康宁片中根皮苷的加标回收数据
样品的测定:取肾康宁片(正大青春宝药业有限公司生产,批号:1601002),按照前述方法制备供试品溶液,精密吸取对照品溶液和供试品溶液各20μl,注入高效液相色谱仪测定,记录各成分的峰面积,外标法定量,肾康宁片中中各含量的测定结果见表21。
表21肾康宁片中各成分的含量
实施例3锁阳指纹图谱的制备
本实施例与实施例1的区别在于高效液相法测定后的数据处理方法不同,对照品和供试品的制备方法相同。
测定法:分别精密吸取对照品溶液和供试品溶液各5μl,注入高效液相色谱仪测定。采用“中药色谱指纹图谱相似度评价系统2004a版”对多批锁阳药材指纹图谱进行数据处理。
空白试验:本实施例的空白试验与实施例1相同,提取用的溶剂纯水在样品色谱图中没有色谱峰,避免了指纹图谱制备中溶剂峰的影响。稳定性试验:称取6号锁阳样品(产自甘肃省肃南县)约2g,精密称定后,按照前述方法制备供试品溶液,将此供试品溶液在0h、2h、4h、6h、8h、10h、12h、16h、20h、24h、30h、36h、42h、48h进液相色谱仪测定,用“中药色谱指纹图谱相似度评价系统2004a版”计算各次进样的相似度。如表22所示,各次进样图谱的相似度均在0.994以上,表明供试品在测定的48h内保持稳定。另外根据各次进样的图谱可见60min后已没有特征峰出现,所以确定指纹图谱的记录时间为60min。
表22供试品溶液稳定性相似度评价结果
精密度试验:称取6号锁阳样品(产自甘肃省肃南县)约2g,精密称定后,按照前述方法制备供试品溶液,将此供试品溶液连续进样5次,考察进样精密度,用“中药色谱指纹图谱相似度评价系统2004a版”计算各次进样的相似度。如表23所示,5次进样图谱的相似度均在0.994以上,表明供试品进样精密度良好。
表23精密度试验相似度评价结果
重现性试验:称取6号锁阳样品(产自甘肃省肃南县)约2g,平行制备5份供试品溶液,将5份供试品溶液进高效液相色谱仪测定,用“中药色谱指纹图谱相似度评价系统2004a版”计算各平行样的相似度。5份平行样品图谱相似度见表24,均在0.996以上,表明本法制备指纹图谱的重现性良好。
表24重现性试验相似度评价结果
锁阳指纹图谱的制备:取18批锁阳样品,按照前述供试品溶液制备方法制备供试品溶液,精密吸取对照品溶液及各供试品溶液各5μl,注入高效液相色谱仪,记录对照品溶液及18批锁阳供试品溶液的色谱图,用“中药色谱指纹图谱相似度评价系统2004a版”处理各色谱图,得到如图9、10所示的指纹图谱及表25所示的相似度评价结果。结果分析,采自不同地域的锁阳样品,由于其土壤等生长环境的影响,其中的化合物组成会有差异。
表25相似度评价结果
在详细说明的较佳实施例之后,熟悉该项技术人士可清楚地了解,在不脱离上述申请专利范围与精神下可进行各种变化与修改,凡依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均属于本发明技术方案的范围。且本发明亦不受限于说明书中所举实例的实施方式。
参考文献
[1]马建滨,都玉蓉.锁阳的化学成分及药理作用[j].青海师范大学学报:自然科学版,2008,2008(2):72-75.
[2]崔树德,等.中药大全[m].哈尔滨:黑龙江科学技术出版社:1997:574-575.
[3]中国药典,2005年版[s].一部.2005:241.
[4]聂莉莎,马丽杰,爱民.锁阳药性及功能的现代研究进展[j].中国民族民间医药,2009,18(16):17-18.
[5]齐艳华,苏格尔.锁阳的研究进展[j].中草药,2000,31(2):146-148.
[6]陈圆华,谢志兵,董静洲.锁阳综合研究概况[j].经济林研究,2005,23(4):114-117.
[7]苏格尔,常艳旭.锁阳的化学成分及药理作用研究概况[j].中国民族医药杂志,2005,11(6):46-49.
[8]陶晶,徐文豪.锁阳茎的化学成分及其药理活性研究[j].中国中药杂志,1999,24(5):292-294.
[9]张遵仪,韩鹏飞,梁满达,等.原儿茶酸衍生物的化学结构与冠脉流量、心肌耗氧量关系的探讨[j].药学学报,1980(11).
[10]李红兵,刘晔玮,李立,等.锁阳清除自由基活性的研究[j].食品科技,2009,34(10):166-169.
[11]王勤,孙芸.hplc法测定锁阳中没食子酸和原儿茶酸的含量[j].西北药学杂志,2010,25(3):190-192.
[12]杨瀚春,王荣,夏鹏霄,等.用hplc法同时测定锁阳的多种有效成分[j].药学服务与研究,2014,14(2):122-124.
[13]李辰,陈卫林,郭玫,等.锁阳中儿茶素、根皮苷的提取及含量测定[j]五邑大学学报(自然科学版),2011,25(1):23-28.
[14]刘晔玮,陈卫林,郭玫,等.锁阳高效液相指纹图谱研究[j].时珍国医国药,2011,22(6):1367-1369.
- 还没有人留言评论。精彩留言会获得点赞!