一种利用固体核磁碳谱检测煤结构参数的定量分析方法与流程
本发明涉及固体核磁碳谱分析技术领域,特别涉及一种利用固体核磁碳谱检测煤结构参数的定量分析方法。
背景技术:
煤是一种由多种官能团、多种化学键组成的复杂有机大分子。了解煤大分子结构模型对认识煤的物理化学性质有重要意义。从煤有机分子的碳结构角度,可以揭示煤液化产物的碳结构变化,为推导煤液化反应机理奠定良好的基础。对煤组成结构深入研究,与工艺性能相结合,能更好的指导工业生产,实现煤炭清洁高效的利用。
煤中只有少部分是可溶于各种溶剂的小分子化合物,其余的大部分是不能被溶解的大分子骨架结构。运用固体核磁共振技术可以在对煤进行非破坏性研究情况下,直接检测煤样,得到煤碳结构参数。由于13c核天然丰度低,13c的nmr信号弱、探测灵敏度低,而且由于外磁场中核的各种相互作用以及固体中化学位移各向异性,磁核之间的直接偶极相互作用引起了谱线增宽、不对称线型,分辨率下降情况。直到70年代中期,随着固体核磁交叉极化(cp)和魔角旋转(mas)等技术发展,固体13c-nmr逐渐应用于煤的研究中。cp技术增强了稀核信号,提高灵敏度,解决碳原子的纵向弛豫时间(t1)太长的问题;而mas技术则消除化学位移的各向异性;边带压制技术(toss)消除样品快速旋转时使得某些原子核的共振谱线产生较大的旋转边带。所以为了窄化谱线、增加灵敏度,得到高分辨率的固体13c-nmr图谱时通常联合使cp、mas和toss等几种技术,这已成为当今研究煤分子结构的普遍方法。
80年代固体核磁技术在常规固体高分辨核磁共振谱的基础上又发展了偶极相移技术,可以区分质子化碳和非质子化碳,提供芳碳率、芳氢率及脂碳率等新的结构信息。到了90年代,利用常规固体技术和偶极相移技术结合谱图分段积分方法得到更为详细的碳结构参数,但是偶极相移技术操作较复杂,需要多次实验,耗时甚多。1996年kohkidena等人运用cp和spe13c-nmr测试pm煤碳结构,通过分峰拟合(拟合软件为macalice)的方法将碳信号分为11类含碳官能团信号,如表1所示。
表113cnmr中不同类型碳对应的化学位移
如今基于不同类型碳的不同化学位移归属,结合计算机辅助技术—分峰拟合技术能够直观、精细和快速的得到多种不同类型碳的含量,得到不同碳材料的结构参数。
由于13ccp/toss/masnmr中cp技术将丰核(1h)较大的自旋状态极化转移给较弱的稀核(13c),使稀核(13c)极化而迅速恢复平衡,缩短了测试时间,但在对氢去耦过程中增强了碳原子能量,使得碳谱谱线增强。简而言之,当分子内两个磁核之间空间位置相近时,对氢核去耦时达到饱和的氢核会将能量转移到碳核上,从而使得碳谱谱线增强,该现象称为碳核overhause效应(noe)。因此13ccp/toss/masnmr谱图中碳原子谱线的强度并不能定量的反映分子内不同化学环境下碳原子的相对数量,交叉极化实验中,接触时间的不足、射频场的不均匀性、noe效应的存在都使得固体核磁定量不准确,与理论碳结构参数存在误差,不能准确进行碳材料的定量研究。液体核磁中运用门控去耦技术已消除了核overhause效应,可以很好的进行碳结构定量。而固体核磁定量大多是通过对谱仪硬件的提升以及脉冲序列的巧妙设计,以达到定量效果,但还未见到快速、有效方便的定量方法。90年代robertv.law等在研究中就表明cp技术的运用主要使季碳芳香碳的磁化比例比质子化碳低,所测得的芳香度要偏低。所以对于大量不带质子碳原子的高成熟煤样,如无烟煤测出的误差要相对小一些,而对于大量带质子碳原子的低阶煤中误差就比较大。所以煤结构分析中煤固体核磁碳结构参数定量分析就显得尤为重要。
煤的组成结构模型一直是煤化学研究的核心问题之一。在煤结构方面,中国专利cn104091504a蔺华林等人通过对煤样固体核磁表征以及对煤液化油气质联用分析构建了煤大分子模型。通过固体核磁碳谱测试表征煤的详细碳结构参数,能够为煤结构模型准确构建奠定坚实的基础,所以获得固体核磁碳结构参数的准确合理性就显得尤为重要。由于核overhause效应等因素的存在,运用13ccp/toss/masnmr测试结果对碳材料直接分析,表征碳结构参数不够准确,测定的参数存在一定的误差。
技术实现要素:
本发明的目的在于修正煤中固体核磁碳谱测定碳结构参数的误差,得到相对准确的碳结构参数,提供了一种利用固体核磁碳谱检测煤结构参数的定量分析方法。
所述方法包括如下步骤:
s1)选取模型化合物;
所述模型化合物包括一系列带脂肪侧链和/或含杂原子官能团的固体芳香化合物,各所述模型化合物的芳香度不同,所述芳香度为不饱和碳原子数与总碳原子数之比;
s2)测定各模型化合物的固体核磁碳谱;
s3)建立校正固体核磁碳谱测试误差的回归曲线方程;
根据不同类型碳原子位移归属和步骤s2测定的固体核磁碳谱,运用分峰拟合方法拟合得到各模型化合物的不同类型碳原子含量拟合值,求和分别计算出各模型化合物的饱和碳原子含量拟合值x%和不饱和碳原子含量拟合值y%;
再将各模型化合物的饱和碳原子含量拟合值x%分别与各模型化合物的饱和碳原子含量理论值进行回归分析,获得饱和碳校正固体核磁测试误差的回归曲线方程(ⅰ),
x’=f(x)(ⅰ);
将各模型化合物的不饱和碳原子含量拟合值y%分别与各模型化合物的不饱和碳原子含量理论值进行回归分析,获得不饱和碳校正固体核磁测试误差的回归曲线方程(ⅱ),
y’=f(y)(ⅱ);
其中x’、y’分别为与x和y对应的修正值;
s4)验证回归曲线方程准确性;
将已知结构的验证化合物按照步骤s2相同的测试条件测定固体核磁碳谱,所述验证化合物也为带脂肪侧链和/或含杂原子官能团的固体芳香化合物;同样通过分峰拟合得到验证化合物的不同类型碳原子含量拟合值,求和分别计算出验证化合物的饱和碳原子含量拟合值x%和不饱和碳原子含量拟合值y%,然后分别代入回归曲线方程(ⅰ)和(ⅱ)得到对应的修正值x’和y’,再将修正值与验证化合物的理论值比较,验证回归曲线方程的准确性;若验证化合物的饱和碳原子含量和不饱和碳原子含量的修正值与理论值相对误差大于10%,说明准确性不高,重新调整模型化合物的种类和数量,重复步骤s1~s3,直到验证化合物的饱和碳原子含量和不饱和碳原子含量的修正值与理论值相对误差小于10%;
s5)待测煤样碳结构参数的测定及修正;
将待测煤样按照步骤s2中相同的测试条件进行固体核磁碳谱测试,同样通过分峰拟合得到待测煤样的不同类型碳原子含量拟合值,求和分别计算出待测煤样的饱和碳原子含量拟合值x%和不饱和碳原子含量拟合值y%,然后分别代入回归方程(ⅰ)和(ⅱ)得到待测煤样对应的修正值x’和y’,再根据x’和待测煤样的各个类型的饱和碳原子含量拟合值等比例计算出各个类型的饱和碳原子含量的修正值,根据y’和待测煤样的各个类型的不饱和碳原子含量拟合值等比例计算出各个类型的不饱和碳原子含量的修正值。上述修正值即为修正后的待测煤样碳结构参数。
所述饱和碳原子含量指饱和碳原子(本文中又称脂肪碳)在总的碳原子中的占比,不饱和碳原子含量指不饱和碳原子(本文中又称芳香碳)在总的碳原子中的占比,下同。
优选的,所述模型化合物选自苯系、萘系和蒽菲系化合物中的至少三种化合物,各化合物纯度大于98%,其中芳香度最低的为30~40%,最高的为90~95%。
优选的,所述模型化合物包括:3,4-二甲基苯甲酸、十二烷基苯磺酸钠、2-萘乙酸、2-甲基萘和9-甲基蒽。
优选的,所述验证化合物也选自苯系、萘系和蒽菲系化合物。
优选的,所述验证化合物为9,10-二甲基蒽。
优选的,步骤s2、s4和s5中,模型化合物、验证化合物及待测煤样在测定固体核磁碳谱前,粉碎研磨至80目以下并在65℃下真空干燥24h。
优选的,步骤s2、s4和s5中,固体核磁碳谱测试条件为:脉冲序列为cp/toss,13c共振频率与仪器相匹配,交叉极化接触时间为1~5ms,循环延迟时间为1~10s,魔角转速为3~7khz,转子外径为4~7mm。
优选的,步骤s3中,选用origin软件进行模型化合物非性回归分析,曲线的相关系数r2大于0.95后得到相应的回归曲线方程。
本发明的一些较佳实施例中,回归曲线方程(ⅰ)和(ⅱ)均为非线性二次函数,通式如下:
x’=a1+b1x+c1x2
y’=a2+b2y+c2y2
式中:a1,b1,c1,a2,b2,c2为曲线回归系数。
本发明具有以下优点和有益效果:
本发明的所述方法以一系列带脂肪侧链和/或含杂原子官能团的苯系、萘系和蒽菲系化合物作为模型化合物,通过测定模型化合物的固体核磁碳谱,确定不同模型化合物碳结构参数误差,将模型化合物碳谱分峰拟合的脂肪碳和芳香碳拟合值与样品碳结构的理论值进行回归分析,得到脂肪碳和芳香碳的校正固体核磁碳谱测试误差回归曲线方程,同时再运用已知结构的模型化合物验证回归方程的准确性;通过对不同模型化合物的固体核磁测定及回归分析,利用回归曲线方程对待测煤样的拟合参数进行修正,可有效解决固体核磁碳谱测试中碳结构参数的误差,实现固体核磁碳谱定量分析。本发明的所述方法为13ccp/toss/masnmr技术与非线性回归方程相结合,能够快速方便的获得相对准确的不同类型碳结构参数,为煤中碳结构分析提供了新的技术保障,提供了一种简便易行的结构参数修正方法。应用所述方法能够相对准确的测定煤结构参数,从而可以从有机碳角度更好的了解煤的结构和性质,为煤结构的解析提供新的技术支撑,对煤的高效转化和利用起指导作用。
所述的方法不仅适用于煤中碳结构的分析,同样适用于油页岩矿产类含碳固体物质及生物质类含碳固体物质中的固体核磁碳谱的分析。
附图说明
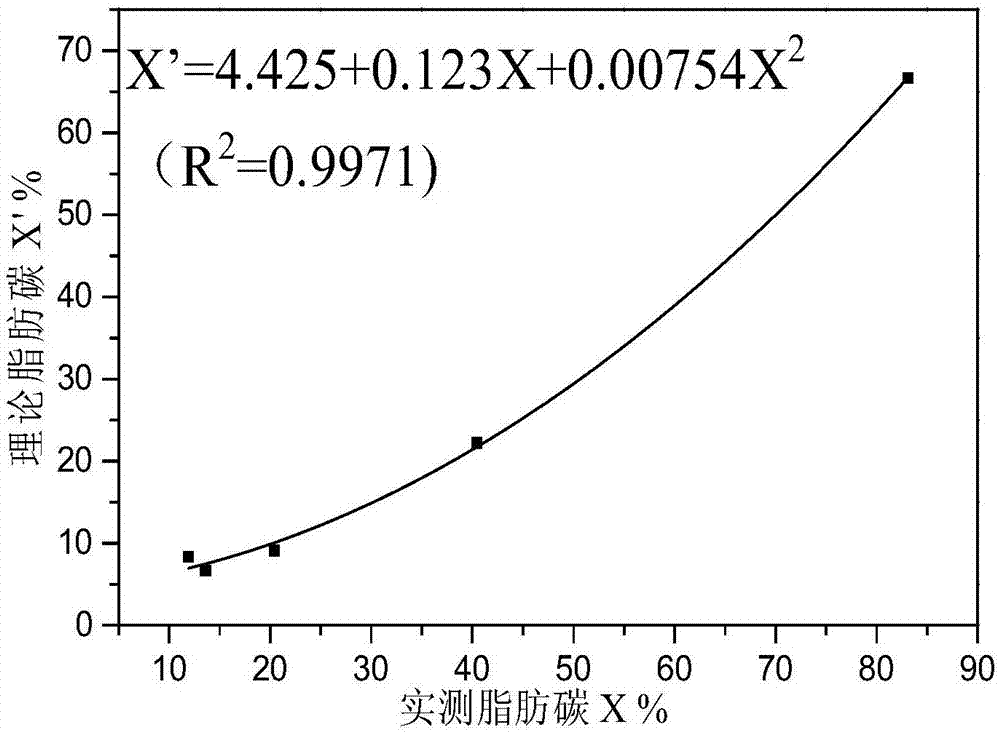
图1为不同模型化合物脂肪碳拟合值与理论值回归曲线图;
图2为不同模型化合物芳香碳拟合值与理论值回归曲线图;
图3为淖毛湖褐煤的分峰拟合图;
图4为小龙潭褐煤的分峰拟合图;
图5为黑山次烟煤的分峰拟合图。
具体实施方式
以下结合具体实施例,对本发明作进一步说明。应理解,以下实施例仅用于本发明而非用于限定本发明的范围。
实施例1新疆淖毛湖褐煤(nmh)碳结构参数的分析
s1)样品及预处理
待测煤样:新疆淖毛湖褐煤。
模型化合物:3,4-二甲基苯甲酸、十二烷基苯磺酸钠、2-萘乙酸、2-甲基萘、9-甲基蒽;如表2所示,其中十二烷基苯磺酸钠的芳香度最低为33.33%,9-甲基蒽的芳香度最高为93.33%,其余的间于两者之间。
验证化合物:9,10-二甲基蒽,芳香度为87.5%。
预处理:将上述待测煤样、模型化合物和验证化合物分别粉碎研磨至80目以下,煤样在65℃下真空干燥24h,干燥、均匀稳定的样品能保证样品在高速旋转或受到强电磁辐射时不爆炸。
s2)测定各模型化合物的固体核磁碳谱
分别将约150mg研磨均匀后的3,4-二甲基苯甲酸、十二烷基苯磺酸钠、2-萘乙酸、2-甲基萘、9-甲基蒽分别装入4mmzro2转子中,在brukeravanceiii500型核磁共振波谱仪上选用脉冲序列为cp/toss进行固体核磁碳谱测试,选用4mm固体高分辨率魔角旋转探头。测试条件为:13c共振频率125.77mhz,交叉极化接触时间为1ms,循环延迟时间为3s,魔角转速为5.6khz,转速为5600r/s。
s3)建立校正固体核磁碳谱测试误差的回归曲线方程
根据表1所示的不同类型碳位移归属和步骤s2测定的固体核磁碳谱,运用分峰拟合方法拟合得到各模型化合物的各个类型碳原子含量的拟合值(x3%,xa%,x2%,x1+x*%,xo%,yh%,yb%,ys%,yo%,ycc1%,ycc2%),求和分别计算出各模型化合物的脂肪碳含量拟合值x%和芳香碳含量拟合值y%,
x=x3+xa+x2+x1+x*+xo,y=yh+yb+ys+yo+ycc1+ycc2。
将各模型化合物的芳香碳和脂肪碳的拟合值与其理论值进行误差比较,结果见表2所示。
表2不同模型化合物碳结构理论值与实测值误差
由表2可知,通过上述常规的测定固定核磁碳谱以及分峰拟合的方法测得的碳结构拟合值与理论值误差较大,原因就是由于核overhause效应等因素使得芳香碳和脂肪碳谱线强度增量不一致。
将步骤s2)中分峰拟合得到的各模型化合物的脂肪碳和芳香碳含量拟合值与各模型化合物的脂肪碳和芳香碳含量理论值进行回归分析,分析结果见图1和图2,相应的,得到脂肪碳校正固体核磁碳谱测试误差非线性回归曲线方程:
x’=4.425+0.123x+0.00754x2(r2=0.9971,n=2)(ⅰa)
以及芳香碳校正固体核磁碳谱测试误差非线性回归曲线方程:
y’=7.861+1.6313y-0.00754y2(r2=0.9971,n=2)(ⅱa)
式中x’、y’分别为与x和y对应的修正值。
脂肪碳的理论值与拟合值以及芳香碳的理论值和拟合值有良好的相关性,相关系数r2=0.9971;换言之固体核磁碳谱测试误差可以用响应非线性二次函数进行修正。。
s4)回归曲线方程准确性验证
将9,10-二甲基蒽按照步骤s2中相同的测试条件测定固体核磁碳谱,分峰拟合得到9,10-二甲基蒽的不同类型碳原子含量拟合值,求和分别计算出9,10-二甲基蒽的饱和碳原子含量和饱和不碳原子含量的拟合值x和y,然后分别代入回归曲线方程(ⅰa)和(ⅱa)得到的x’和y’为修正值,将9,10-二甲基蒽的饱和碳原子含量和不饱和碳原子含量修正前后的拟合值与理论值比较,结果见表3,修正后脂肪碳和芳香碳与其理论值相对误差均在10%以内,故运用非线性回归方法可以很好的修正固体核磁碳谱测试中的脂肪碳和芳香碳含量误差,可以得到较为准确的结构参数。
表39,10-二甲基蒽中碳结构实测值和理论值误差
s5)淖毛湖褐煤(nmh)结构参数的测定及修正
将淖毛湖褐煤煤样按照步骤s2中相同的测试条件进行固体核磁碳谱测试,同样通过分峰拟合得到淖毛湖褐煤煤样的不同类型碳原子含量拟合值,求和分别计算出淖毛湖褐煤煤样的饱和碳原子含量拟合值x%和不饱和碳原子含量拟合值y%,然后分别代入回归方程(ⅰa)和(ⅱa)得到淖毛湖褐煤煤样对应的修正值x’和y’,再根据x’和淖毛湖褐煤煤样的各个类型的饱和碳原子含量拟合值等比例计算出各个类型的饱和碳原子含量的修正值,结果见表4,根据y’和待测煤样的各个类型的不饱和碳原子含量拟合值等比例计算出各个类型的不饱和碳原子含量的修正值,结果见表5。
表4修正前后淖毛湖褐煤不同类型脂肪碳分布
表5修正前后淖毛湖褐煤不同类型芳香碳分布
同时对煤样作元素分析,元素分析结果如表6所示。
以h/c作为对比参数,煤结构中氢原子以脂肪氢和芳香氢的形式存在,其中脂肪氢部分包括甲基、次甲基以及亚甲基形式,而芳香氢中则主要以质子化氢以及羧基中的氢存在,不考虑酚类,则煤的h/c原子比可根据公式估算:h/c=(yh+(1-y)×2.0+ycc1)/100。修正前后淖毛湖褐煤h/c原子比及芳香度(芳香碳含量)见表6。
表6修正前后淖毛湖褐煤h/c及芳香度
由表6可知,运用非线性回归曲线方程对nmh煤修正后的不同结构参数估算的h/c原子比与元素分析结果具有一致性。对nmh煤的不同类型碳结构参数修正可靠,非线性回归曲线方程修正后能够得到相对准确的碳结构参数。
实施例2小龙潭褐煤(xlt)碳结构参数的分析
按照实施例1的分析步骤对小龙潭褐煤进行分析
步骤s5中,将小龙潭褐煤煤样按照步骤s2中相同的测试条件进行固体核磁碳谱测试,同样通过分峰拟合得到小龙潭褐煤煤样的不同类型碳原子含量拟合值,求和分别计算出小龙潭褐煤煤样的饱和碳原子含量拟合值x%和不饱和碳原子含量拟合值y%,然后分别代入回归方程(ⅰa)和(ⅱa)得到小龙潭褐煤煤样对应的修正值x’和y’,再根据x’和小龙潭褐煤煤样的各个类型的饱和碳原子含量拟合值等比例计算出各个类型的饱和碳原子含量的修正值,结果见表7,根据y’和小龙潭褐煤煤样的各个类型的不饱和碳原子含量拟合值等比例计算出各个类型的不饱和碳原子含量的修正值,结果见表8。
表7修正前后小龙潭煤不同类型脂肪碳分布
表8修正前后小龙潭煤不同类型芳香碳分布
同样对小龙潭褐煤进行元素分析,元素分析结果以及修正前后小龙潭煤的h/c原子比及芳香度(芳香碳含量)见表9。
表9修正前后小龙潭煤h/c及芳香度
由表9可知,运用非线性回归曲线方程对xlt煤修正后的不同结构参数估算的h/c原子比与元素分析结果具有一致性。对xlt煤的不同类型碳结构参数修正可靠,非线性回归曲线方程修正后能够得到相对准确的碳结构参数。
实施例3黑山次烟煤(hs)碳结构参数的分析
按照实施例1的分析步骤对黑山次烟煤进行分析
步骤s5中,将黑山次烟煤煤样按照步骤s2中相同的测试条件进行固体核磁碳谱测试,同样通过分峰拟合得到黑山次烟煤煤样的不同类型碳原子含量拟合值,求和分别计算出黑山次烟煤煤样的饱和碳原子含量拟合值x%和不饱和碳原子含量拟合值y%,然后分别代入回归方程(ⅰa)和(ⅱa)得到黑山次烟煤煤样对应的修正值x’和y’,再根据x’和黑山次烟煤煤样的各个类型的饱和碳原子含量拟合值等比例计算出各个类型的饱和碳原子含量的修正值,结果见表10,根据y’和黑山次烟煤煤样的各个类型的不饱和碳原子含量拟合值等比例计算出各个类型的不饱和碳原子含量的修正值,结果见表11。
表10修正前后黑山次烟煤不同类型脂肪碳分布
表11修正前后黑山次烟煤不同类型芳香碳分布
同样对黑山次烟煤煤进行元素分析,元素分析结果以及修正前后黑山次烟煤的h/c原子比及芳香度(芳香碳含量)见表12。
表12修正前后黑山次烟煤h/c及芳香度
由表12可知,运用非线性回归曲线方程对hs煤修正后的不同结构参数估算的h/c原子比与元素分析结果具有一致性。对hs煤的不同类型碳结构参数修正可靠,非线性回归曲线方程修正后能够得到相对准确的碳结构参数。
综上所述,本发明通过测定不同模型化合物固体核磁碳谱测试误差,将不同模型化合物碳谱分峰拟合的脂肪碳和芳香碳测试值与样品碳结构的理论值进行回归分析,得到脂肪碳和芳香碳的校正固体核磁碳谱测试误差的回归曲线方程,同时再运用已知结构的模型化合物验证回归方程的准确性;通过对不同模型化合物的固体核磁测定及回归分析,解决了由于核overhause效应等因素引起的碳结构参数的误差,13ccp/toss/masnmr技术与回归曲线方程相结合能够快速方便的获得相对准确的不同类型碳结构参数,为煤碳结构分析提供了新的技术保障,提供了一种简便易行的结构参数修正方法。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
- 还没有人留言评论。精彩留言会获得点赞!