一种郁金药材挥发性成分一测多评测定方法与流程
本发明属于药物检测领域,具体涉及一种郁金药材挥发性成分一测多评测定方法,所述的方法包含5种倍半萜类成分的含量测定方法。
背景技术:
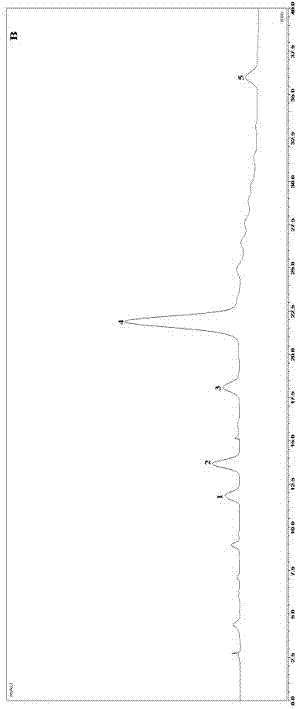
:由于自然界植物中化学成分的复杂性,单一指标成分无法准确的反应植物药的有效成分;且分别制定单一成分的含量测定,在测定方法实施过程中,对照品消耗量大,检测周期长、耗时耗力,效率低。通过研究发现,不同物质在紫外检测器中的响应与其浓度的比值是一个常数,该常数称为相对校正因子。使用a对照品(易获得)外标法测定a化合物的峰面积,通过峰面积及a对照品浓度计算其含量;同时获得的b化合物的峰面积,通过校正因子计算获得b化合物的含量。该替代对照品法被命名为一测多评法。由于大大减少了对照品的用量,尤其是难获得的对照品用量,并缩短了检测周期,一测多评的质量控制方法得到迅速的推广和应用。中国药典也已采纳该方法,对植物来源药物进行多指标含量测定,且数目仍在不断增加。郁金,具有活血止痛,行气解郁,清心凉血,利胆退黄的功效,在安宫牛黄丸、十香返生丸、牛黄清宫丸、醒脑静注射液等均有使用。中国药典一部规定的郁金基原为三种,主要产地为广西、广东和四川。挥发油是中药郁金的主要活性成分,含量约为0.3%左右,多为倍半萜类化合物。单个含量测定的成分选择,将严重影响郁金成分测定方法的可靠性和实用性。本发明对此进行了研究,找到了一种对郁金中5种倍半萜类成分进行检测的方法,从而解决了相关的技术问题。技术实现要素:本发明的目的在于提供一种郁金中5种倍半萜类成分的含量测定方法。本发明的检测方法,可以从源头药材进行质量控制,探讨符合制剂要求的检测标准,从而保证最终产品质量的安全性、稳定性和有效性,达到快速、高效质量控制的目的。本发明提供一种郁金中5种倍半萜类成分的含量测定方法,其特征在于,步骤如下:(1)供试品溶液的制备:取郁金药材,用水蒸气蒸馏法提取挥发油,过c8或c18固定相柱,流出液弃去,用甲醇洗脱,收集甲醇洗脱液定容,作为供试品溶液;(2)对照品溶液的制备;取莪术二酮,用甲醇溶解,定容,作为对照品溶液;(3)取供试品溶液和对照品溶液,分别注入高效液相色谱仪,得到色谱图;(4)先以对照品莪术二酮为参照,用外标一点法或工作曲线法计算供试品溶液中莪术二酮含量,再使用校正因子计算供试品溶液中姜黄烯酮,莪术烯醇,莪术酮及吉马酮的含量。其中,步骤(1)方法如下:取粉碎过筛的郁金30g,加水1000-1500ml,提取芳香水500-1000ml,混匀,精密吸取5-10ml,通过c8或c18固相萃取小柱,流出液弃去,用1-10ml甲醇洗脱,收集洗脱液定容至1-10ml,摇匀,作为供试品溶液。其中,步骤(2)方法如下:取莪术二酮适量,精密称定,用甲醇溶解,定容,如果使用外标法,则使用浓度为50μg/ml莪术二酮对照品溶液,如果使用工作曲线法,则采用浓度范围1-100μg/ml的至少五个梯度的莪术二酮对照品溶液。其中,步骤(3)的色谱条件如下:采用c18色谱柱;流动相为乙腈(a)-0.1%甲酸水溶液(b)梯度洗脱浓度3种任选1)0~18min,58%a;18~25min,68%a;25~40min,68%a;2)0~15min,58%a;15~25min,68%a;25~40min,78%a3)0~20min,48%a;20~30min,68%a;30~50min,78%a进样量10-20μl;流速0.1-1ml·min-1;柱温35℃;检测波长254nm;210nm;其中,步骤(4)方法如下:先用外标法或工作曲线法计算供试品溶液中莪术二酮含量,再通过公式(1)计算其余4种成分的含量:公式中(1)中,cs为被测物含量as为被测物峰面积ci为莪术二酮含量ai为莪术二酮峰面积fs/i为校正因子。优选的,其中,步骤(1)方法如下:取粉碎过筛的郁金30g,加水1500ml,提取芳香水1000ml,混匀,精密吸取5ml,通过c8固相萃取小柱,流出液弃去,用1ml甲醇洗脱,收集洗脱液定容至1ml,摇匀,作为供试品溶液。其中,步骤(3)色谱条件如下:c18色谱柱;流动相乙腈(a)-0.1%甲酸水溶液(b)0~18min,58%a;18~25min,68%a;25~40min,68%a;进样量20μl;流速0.3ml·min-1;柱温35℃;检测波长0~15min,254nm;15~40min,210nm。本发明的检测方法是经过筛选获得的,筛选过程如下:1、专属性与阴性、空白比较无干扰2、线性关系考察取混合对照品溶液,分别进样,以测得的峰面积为纵坐标,对应溶液的浓度为横坐标,进行线性回归分析,得到各对照品的回归方程及线性范围。表1线性关系考察结果组分回归方程r值姜黄烯酮y=1.2x-0.20.9999莪术烯醇y=0.8x-0.20.9998莪术二酮y=0.6x-0.20.9999莪术醇y=2.5x-3.40.9996吉马酮y=2.5x+0.00.99973、精密度试验取混合对照品溶液,于同一日内连续进样6次,测定溶液中姜黄烯酮、莪术烯醇、莪术二酮、莪术酮及吉马酮的峰面积并计算rsd值。结果分别为0.7%、0.5%、0.9%、0.3%及0.8%,表明仪器日内精密度良好。4、重复性试验平行制备6份供试品溶液,分别进样,测定姜黄烯酮、莪术烯醇、莪术二酮、莪术酮及吉马酮含量,并计算rsd值。含量的rsd值分别为1.0%、2.1%、0.9%、0.3%及1.2%,表明该方法的重复性良好。5、稳定性试验供试品溶液制备后第0、2、4、8、12、24h进样,测定供试品溶液中姜黄烯酮、莪术烯醇、莪术二酮、莪术酮及吉马酮的峰面积,并计算rsd值。结果供试品溶液中,上述成分峰面积的rsd值分别为1.5%、1.3%、0.7%、0.7%及2.1%,表明供试品溶液在24小时内的稳定性良好。6、加样回收试验按照样品量和对照品加入量大约为0.8,1.0和1.2的比例,精密加入对照品混标溶液(混标溶液,供试品溶液色谱条件进行分析,计算供试品溶液中5个待测成分的平均加样回收率及rsd值,结果下表。表2加样回收试验结果7、相对校正因子的确定按色谱条件进行检测,记录5个目标化合物的峰面积,以莪术二酮为内参物,按公式(1)多点法计算其余4个成分对莪术二酮的相对校正因子,结果姜黄烯酮、莪术烯醇、莪术酮、吉马酮对莪术二酮的相对校正因子。as和cs分别为待测成分的峰面积和对应的浓度;ai和ci分别为内参物的峰面积和对应的浓度。表3相对校正因子的测定结果a:姜黄烯酮对莪术二酮的相对校正因子;b:莪术烯醇对莪术二酮的相对校正因子;c:莪术酮对莪术二酮的相对校正因子;d:吉马酮对莪术二酮的相对校正因子。本发明的检测方法与现有检测方法相比较,具有以下优点:目前,尚未有针对郁金挥发油成分的一测多评多指标含量测定方法,本发明开发的方法操作简便、准确可靠,可作郁金倍半萜类成分的检测手段。使用廉价易得的莪术二酮,同时测定5种倍半萜类成分,节约生产成本,提高生产效率和经济效益,方法快速、准确,能更全面的覆盖药材质量。附图说明图1、对照品图谱(1.姜黄烯酮2.莪术烯醇3.莪术二酮4.莪术酮5.吉马酮)图2、5种倍半萜类紫外吸收光谱图(1.姜黄烯酮2.莪术烯醇3.莪术二酮4.莪术酮5.吉马酮)具体实施方式通过以下具体实施例对本发明做进一步的说明,但不作为限制。实施例1步骤(1)供试品溶液制备方法如下:取粉碎过筛的郁金30g,加水1000ml,提取芳香水500ml,混匀,精密吸取5ml,通过c18固相萃取小柱,流出液弃去,用1ml甲醇洗脱,收集洗脱液定容至1ml,摇匀,作为供试品溶液。步骤(2)对照品溶液的制备:取莪术二酮适量,精密称定,使用外标一点法或工作曲线法计算莪术二酮含量;其中外标法使用浓度为50μg/ml莪术二酮对照品溶液。步骤(3)色谱条件与系统适用性试验:照高效液相色谱测定法;c18色谱柱;流动相乙腈(a)-0.1%甲酸水溶液(b)梯度洗脱浓度3种任选0~18min,58%a;18~25min,68%a;25~40min,68%a;进样量10μl;流速0.6ml·min-1;柱温35℃;检测波长254nm;210nm。其中,步骤(4)方法如下:通过公式(1)计算其余4种成分的含量并与外标法实际测定结果进行比较;公式中(1)中,cs为被测物含量as为被测物峰面积ci为莪术二酮含量ai为莪术二酮峰面积fs/i为校正因子计算获得5种倍半萜类含量如下:实施例2步骤(1)供试品溶液制备方法如下:取粉碎过筛的郁金30g,加水1500ml,提取芳香水500ml,混匀,精密吸取10ml,通过c8固相萃取小柱,流出液弃去,用10ml甲醇洗脱,收集洗脱液定容至10ml,摇匀,作为供试品溶液。步骤(2)对照品溶液的制备:取莪术二酮适量,精密称定,使用外标一点法或工作曲线法计算莪术二酮含量;工作曲线采用浓度范围1-100μg/ml的至少五个梯度的莪术二酮对照品溶液。步骤(3)色谱条件与系统适用性试验:照高效液相色谱测定法;c18色谱柱;流动相乙腈(a)-0.1%甲酸水溶液(b)梯度洗脱浓度3种任选0~15min,58%a;15~25min,68%a;25~40min,78%a进样量20μl;流速0.3ml·min-1;柱温35℃;检测波长254nm;210nm。步骤(4)方法如下:通过公式(1)计算其余4种成分的含量并与外标法实际测定结果进行比较;公式中(1)中,cs为被测物含量as为被测物峰面积ci为莪术二酮含量ai为莪术二酮峰面积fs/i为校正因子计算获得5种倍半萜类含量如下:实施例3步骤(1)供试品溶液的制备方法如下:取粉碎过筛的郁金30g,加水1500ml,提取芳香水1000ml,混匀,精密吸取5ml,通过c8固相萃取小柱,流出液弃去,用1ml甲醇洗脱,收集洗脱液定容至10ml,摇匀,作为供试品溶液。步骤(2)对照品溶液的制备:取莪术二酮适量,精密称定,使用外标一点法或工作曲线法计算莪术二酮含量;其中外标法使用浓度为50μg/ml莪术二酮对照品溶液。步骤(3)色谱条件与系统适用性试验:照高效液相色谱测定法;c18色谱柱;流动相乙腈(a)-0.1%甲酸水溶液(b)梯度洗脱浓度3种任选0~18min,58%a;18~25min,68%a;25~40min,68%a;进样量20μl;流速0.31ml·min-1;柱温35℃;检测波长254nm;210nm。步骤(4)方法如下:通过公式(1)计算其余4种成分的含量并与外标法实际测定结果进行比较;公式中(1)中,cs为被测物含量as为被测物峰面积ci为莪术二酮含量ai为莪术二酮峰面积fs/i为校正因子计算获得5种倍半萜类含量如下:当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1