一种同时定量检测小鼠血浆中黄芪甲苷-IV、环黄芪醇的方法与流程
本发明属于医药领域,具体涉及一种基于uplc-hrms的小鼠血浆中黄芪甲苷-iv、环黄芪醇同时定量方法的建立及药物代谢动力学应用。
背景技术
黄芪甲苷-iv(astragalosideiv,ast)作为中草药黄芪的主要活性成分,它具有多种药理作用,包括抗炎,抗高血压,心脏保护,抗氧化和抗细胞凋亡。据报道ast也是各种代谢综合征的潜在治疗药物。ast主要的具有生物活性的代谢物是环黄芪醇(cycloastragenol,cst),它是一种小分子端粒酶活化剂和潜在的脂肪形成抑制剂。此外,最近的研究表明ast和cst在抑制活性氧(reactiveoxygenspecies,ros)相关的内质网应激和抑制炎症因子等方面具有同等功效,它们可以刺激细胞外信号调节蛋白激酶的磷酸化以提高免疫力,促进体外和体内的伤口愈合。尽管对于ast和cst有大量的药效作用研究,他们的药理作用关系尚未明确。因此,我们仍需要利用两种化合物的定量方法来研究它们的药物代谢动力学和药效学,并进一步评估ast作为临床治疗药物的可能性。
现有的一些基于液相色谱-质谱(lc-ms或lc-ms/ms)单独定量ast或cst的方法仍然存在一些缺点,如样品前处理过程复杂,检测时间长,所需样品量大或灵敏度低。为了同时测定两种物质,zeng等人建立了一种在10分钟内分析ast和cst的具有较高灵敏度的定量方法,但该方法分析过程中需要正负离子模式切换。另一种方法需要很长的数据采集时间(17分钟)。此外,上述两种方法的样本量均为50μl血浆,并不适合用于小鼠血浆中的定量。另外,由于ast具有较高相对分子质量和较低溶解度,在动物体内的生物利用度并不高。综上所述,我们有必要建立灵敏度更高的方法来同时定量ast和cst。
技术实现要素:
本发明的目的是提供一种检测小鼠血浆中黄芪甲苷-iv、环黄芪醇的方法。
本发明所提供的检测小鼠血浆中黄芪甲苷-iv、环黄芪醇的方法,包括步骤(1)样品的制备和步骤(2)采用uplc-hrms进行检测。
步骤(1)样品的制备采用包括以下步骤的方法:
a)向待测血浆样品中加入内标工作溶液和甲醇,得到含有内标和甲醇的待测血浆;
b)向所述含有内标和甲醇的待测血浆中加入乙腈沉淀蛋白,混匀,离心,收集上清液;
c)向所述上清液中加入甲酸溶液,用去磷脂板过滤,收集滤液,得到待测样品。
步骤a)中,所述内标工作溶液中所述内标为地高辛。
所述内标工作溶液具体通过包括下述步骤的方法配制:精密称取地高辛标准品适量,用甲醇溶解成1mg/ml的标准溶液储备液;用甲醇稀释成浓度为100ng/ml的内标工作溶液。
所述待测血浆样品与内标工作溶液和甲醇的体积比依次可为:2:1:1。
步骤b)中,乙腈与所述待测血浆样品的体积比可为15:2。
所述混匀的条件可为:2000rpm涡旋混匀5min。
所述离心的条件可为:4000rpm离心5min。
步骤c)中,所述甲酸溶液为体积浓度10%的甲酸溶液。
所述甲酸溶液与步骤a)中的待测血浆样品的体积比可为:1:1。
步骤(2)检测所用超高效液相色谱条件如下:
色谱柱为:zorbaxextend-c18rrhd;
流动相采用含有0.1%甲酸的水为a相,含有0.1%甲酸的甲醇和乙腈的混合液(甲醇与乙腈的体积比为50:50)为b相;(0.1%甲酸是甲酸相当于水/甲醇乙腈的混合液的体积比);
洗脱方式为梯度洗脱;
所述梯度洗脱的程序如下:
0-0.2min:b相占流动相总体积的25%;
0.2-1.0min:b相的体积分数由25%增加至85%;
1.0-2.0min:b相的体积分数维持85%不变;
2.0min-2.1min:b相的体积分数由85%增加至95%;
2.1min-2.5min:b相的体积分数维持95%不变;
2.5min-2.51min:b相的体积分数由95%减少至25%;
2.51min-3min:b相的体积分数维持25%不变。
步骤(2)检测所用质谱条件如下:采用q-ot-qit杂合型质谱仪,配有esi与apci源,采用正离子模式的esi检测方式,扫描方式为选择离子监测(selectedionmonitoring,sim)。
所述超高效液相色谱条件中,色谱柱具体为zorbaxextend-c18rrhd(2.1×50mm,1.8μm;agilentcorporation,usa)。
流动相流速为0.5ml/min,梯度洗脱总时间为3min,进样量为10μl,柱温为40℃。
所述质谱条件中,其他质谱参数如下:喷雾电压:3800v;鞘气:25;辅助气:15;反吹气0;离子传输管温度:350℃;离子源温度:450℃;质谱分辨率:120000;隔离窗口:1da;最大注入时间:150ms。
所述监测离子质荷比(m/z)如下:黄芪甲苷-iv、环黄芪醇和地高辛主要离子分子为[m+na]+,其质荷比分别为m/z807.4501(ast),m/z513.3550(cst),m/z803.4197(地高辛)。
ast、cst和内标的保留时间分别为1.93min,2.13min,1.66min。
本发明还包括一种同时检测小鼠血浆中黄芪甲苷-iv、环黄芪醇的含量的方法,具体包括下述步骤:
1)标准曲线的制备:取一系列浓度的黄芪甲苷-iv、环黄芪醇的混合标准品溶液加入空白血浆样品中,按照上述的样品制备方法进行制备,然后对得到的上清液按照上述的uplc-hrms法进行检测,并分别记录每个浓度的黄芪甲苷-iv、环黄芪醇对应的峰面积;以黄芪甲苷-iv与内标峰面积比值y为纵坐标,以黄芪甲苷-iv浓度x为横坐标,制备黄芪甲苷-iv的线性回归方程;以环黄芪醇与内标峰面积比值y为纵坐标,以环黄芪醇浓度x为横坐标,制备环黄芪醇的线性回归方程;
2)待测血浆样品中黄芪甲苷-iv、环黄芪醇的含量测定:将待测血浆样品按照上述的样品制备方法进行制备,然后对得到的上清液按照上述的液相色谱串联质谱法进行检测,并分别记录黄芪甲苷-iv、环黄芪醇对应的峰面积;计算黄芪甲苷-iv与内标峰面积比值y,将y值代入所述黄芪甲苷-iv的线性回归方程中,计算得到所述待测血浆样品中黄芪甲苷-iv的浓度;计算环黄芪醇与内标峰面积比值y,将y值代入所述环黄芪醇的线性回归方程中,计算得到所述待测血浆样品中环黄芪醇的浓度。
本发明所述方法可成功用于黄芪甲苷-iv的药代动力学研究。
本发明建立了一种基于超高效液相色谱-高分辨质谱(ultra-high-performanceliquidchromatography–high-resolutionmassspectrometry,uplc-hrms)的快速,灵敏,专属性高的方法,可以在3分钟内同时定量ast和cst。此外,本发明方法只需要20μl血浆进行样品制备。在简单的蛋白质沉淀过程之后,进一步通过去磷脂板过滤样品。在正离子电喷雾电离(electrosprayionization,esi)和选择离子监测(selectedionmonitoring,sim)模式下,在orbitrapfushionlumos杂合型质谱仪(quadrupole–orbitrap–quadrupoleiontrap,q-ot-qit)上进行检测,并成功应用这种方法来定量给药30mg/kg/dast的小鼠血浆中的ast和cst含量。
附图说明
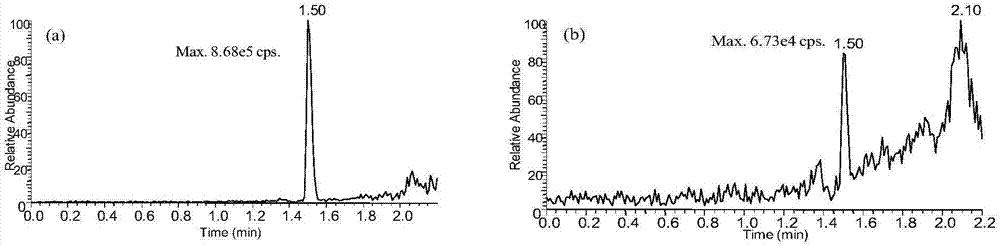
图1为使用(a)和不使用去磷脂板(b)过滤后进样的黄芪甲苷-iv提取离子流色谱图。
图2.不同倍数乙腈作为提取溶剂对黄芪甲苷-iv(ast,100ng/ml),环黄芪醇(cst,100ng/ml)和内标地高辛(digoxin,100ng/ml)的提取效果,(a)1倍乙腈提取结果(b)2倍乙腈提取结果(c)3倍乙腈提取结果(d)5倍乙腈提取结果(e)7.5倍乙腈提取结果。
图3为两种内标与黄芪甲苷的提取离子流色谱图,ast:黄芪甲苷-iv,rg1:人参皂苷rg1,digoxin:地高辛。
图4为黄芪甲苷-iv(a)、环黄芪醇(b)、地高辛(c)在正离子全扫描模式下的质谱图。
图5为黄芪甲苷-iv(a),环黄芪醇(b),地高辛(c)在分辨率为15000(i),30000(ii),60000(iii),120000(iv),240000(v)时的质谱图;其中,黄芪甲苷-iv,环黄芪醇在血浆中的浓度均为1.5ng/ml,地高辛浓度为50ng/ml。
图6为不同分辨率下黄芪甲苷-iv(1.5ng/ml),环黄芪醇(1.5ng/ml)和地高辛(50ng/ml)的响应强度。
图7为黄芪甲苷-iv(i),环黄芪醇(ii),地高辛(iii)在空白血浆(a)、加入1ng/ml标准溶液的血浆(b)及给药30mg/kg/d黄芪甲苷-iv后血浆(c)中的提取离子流色谱图。
图8为黄芪甲苷-iv(ast)与环黄芪醇(cst)的血药浓度-时间曲线。
具体实施方式
下面通过具体实施例对本发明进行说明,但本发明并不局限于此。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、生物材料等,如无特殊说明,均可从商业途径得到。
实施例
1.1实验试剂
黄芪甲苷-iv(astragalosideiv,ast),分子式c41h68o14,相对分子质量784.4609,购于中国药品生物制品检定所;环黄芪醇(cycloastragenol,cst),分子式c30h50o5,相对分子质量490.3658,内标地高辛(digoxin),分子式c41h64o14,相对分子质量780.4296,均购于成都康邦生物科技有限公司,纯度hplc>98%,密封避光保存在2-8℃。乙腈、甲醇、(色谱纯)购自德国merck公司,甲酸(色谱纯)购自美国roe公司。实验用水为娃哈哈纯净水。
2.2血浆样品采集
24只雄性km小鼠(8周龄,体重35-40g)购自北京维通利华实验动物技术有限公司。所有动物在12h光照-黑暗循环,恒温(23±2℃)、恒湿(60%),自由进食饮水条件下适应性饲养一周。每次内眦取血前4h禁食不禁水。动物实验操作遵循中央民族大学实验动物福利和伦理要求。
2.3样品制备
标准品溶液的配制:精密称取黄芪甲苷-iv,环黄芪醇,地高辛标准品适量,分别用甲醇溶解成1mg/ml的标准溶液储备液。取黄芪甲苷-iv和环黄芪醇标准溶液储备液适量,用甲醇稀释成浓度为2,3,10,30,100,200,400ng/ml的标准系列溶液。取地高辛标准溶液储备液适量,用甲醇稀释成浓度为100ng/ml的内标工作溶液。另外精密称取黄芪甲苷-iv和环黄芪醇标准品各一份,用甲醇溶解并稀释成1mg/ml的标准溶液储备液,并稀释成浓度为3,30,300ng/ml的质量控制(qc)工作溶液。
给药溶液的配制:在室温条件下,精密称取羧甲基纤维素钠适量,用水溶解并在磁力搅拌器上搅拌过夜。称取75mg黄芪甲苷-iv,溶解于50ml0.5%羧甲基纤维素钠溶液中,使用磁力搅拌器充分混匀3h,使黄芪甲苷-iv终浓度为1.5mg/ml。
血浆样品前处理:实验前将血浆样本室温解冻,充分混匀后,取空白血浆20μl,加入内标工作溶液10μl,甲醇10μl(或10μl含黄芪甲苷-iv,环黄芪醇的标准溶液或质控溶液),加入150μl乙腈沉淀蛋白,2000rpm涡旋混匀5min,4000rpm离心5min后,在上清液中加入20μl10%的甲酸溶液,随后用去磷脂板过滤。取150μl过滤后的样品待测。
2.4实验仪器与条件
2.4.1实验仪器
移液器(美国thermo公司);离心机与混匀仪(德国eppendorf公司);watersostrotm96-wellplate25mg1/pkg去磷脂板(美国waters公司)。q-ot-qit杂合型质谱仪(orbitrapfusionlumos,美国thermo公司)。
2.4.2lc-ms分析
(1)色谱条件
色谱仪:超高效液相色谱(ultramate3000,美国thermo公司)
色谱柱:zorbaxextend-c18rrhd(2.1×50mm,1.8μm;agilentcorporation,usa)。
流动相:a:水(0.1%甲酸)b:甲醇:乙腈(50:50,v/v)加0.1%甲酸,流速0.5ml/min,梯度洗脱总时间3min,进样量10μl,柱温40℃。
(2)质谱条件
质谱仪:q-ot-qit杂合型质谱仪(orbitrapfusionlumos,美国thermo公司),配有esi与apci源。
采用正离子模式的esi检测方式,扫描方式为选择离子监测(selectedionmonitoring,sim),监测离子质荷比(m/z)如下表所示:
实验中所使用的气体均为氮气。数据采集及处理软件采用xcalibur2.2数据处理系统。其他质谱参数如下:
2.5方法学验证
方法专属性:取空白小鼠血浆样品20μl,按“血浆样品前处理”项下操作,制备空白样品;将一定浓度的黄芪甲苷-iv、环黄芪醇标准溶液和内标地高辛溶液加入空白血浆中,依同法操作,制备空白加标样品。将空白样品和空白加标样品分别进行lc-ms分析,记录相应的色谱图,用于评价方法的专属性。
线性范围与定量下限:取空白小鼠血浆样本20μl,加入黄芪甲苷-iv、环黄芪醇标准系列溶液各10μl,配制成相当于黄芪甲苷-iv、环黄芪醇浓度为1,1.5,5,15,50,100,200ng/ml的血浆样本,按“血浆样品前处理”项下操作,制备工作曲线。以血浆样本中待测物浓度为横坐标,待测物与内标物的峰面积比值为纵坐标,用加权(w=1/x2)最小二乘法进行回归运算,求得的直线回归方程即为工作曲线。分析方法的定量下限(lowerlimitofquantification,lloq)定义为准确度和精密度在±20%范围内的最低浓度。信噪比为3时的待测物浓度定义为方法的最低检出限(limitofdetection,lod)。
准确度和精密度:取空白小鼠血浆样本20μl,加入qc工作溶液各10μl,按“血浆样品前处理”项下操作,制备黄芪甲苷-iv、环黄芪醇低、中、高三个浓度(分别为3,30,300ng/ml)的质量控制样品,每浓度6个样本,连续测定3天,根据当日的工作曲线,得出qc样品的测得浓度。计算qc样本的相对偏差(relativeerror,re)和相对标准偏差(relativestandarddeviation,rsd),分别用于评价方法的准确度和精密度。其中准确度re应在±15%的范围内,日内和日间精密度rsd应<15%。
提取回收率:取空白小鼠血浆样本20μl,按“血浆样品前处理”项下操作,制备低、中、高三个浓度(黄芪甲苷-iv、环黄芪醇浓度分别为3,30,300ng/ml)的样品,每浓度6个重复。另取空白小鼠血浆样本20μl,按“血浆样品前处理”项下操作,在获得的样品中加入低、中、高三个浓度的标准溶液,2000rpm涡旋混合,进行lc-ms分析,获得相应的色谱峰面积(三个平行样本的平均值)。以每一浓度两种处理方式所获得的色谱峰面积的比值计算两种待测物的提取回收率。
基质效应:分别取空白小鼠血浆20μl,按“血浆样品前处理”项下操作,制备低、中、高三个浓度(黄芪甲苷-iv、环黄芪醇浓度分别为3,30,300ng/ml)的样品,每浓度6个重复。另外取等量的水替代小鼠血浆,按“血浆样品前处理”项下操作,制备低、中、高三个浓度(黄芪甲苷-iv、环黄芪醇浓度分别为3,30,300ng/ml)的样品,每浓度6个重复。以每一浓度两种处理方法的色谱峰面积的比值计算两种待测物的基质效应。
稳定性考察:按照“血浆样品前处理”项下操作制备低、中、高三个浓度(黄芪甲苷-iv、环黄芪醇浓度分别为3,30,300ng/ml)的qc样品,分别考察其在-80℃保存一个月,室温保存8h,反复冻融3次,待测物和内标储备液在2℃–8℃保存2周以及样品处理后于室温下在进样小瓶中保存2h的稳定性。通过分析6个加入10μg/ml待测物的小鼠血浆重复样本,并用空白小鼠血浆将其稀释至1.5,15和150ng/ml来探究稀释效应。
2.6药物代谢动力学
所有小鼠给药ast30mg/kg,在给药后20min,40min,1h,1.5h,2h,3h,4h,6h,8h,10h,12h,16h,20h和24h内眦取血约100μl。在接下来的六天中每天灌胃给予ast30mg/kg,以评估重复给药是否会有药物积累作用。每天在给药前采血。用涂有肝素钠的离心管收集血液,4,000rpm离心10min,将血浆保存在-80℃待用。
3结果与讨论
3.1样品前处理条件的优化
根据待测物的结构特点和化学性质,结合文献报道,本研究对于蛋白质沉淀法、液液萃取的方法,使用乙腈、正丁醇、乙醚:二氯甲烷(3:2,v/v),乙酸乙酯有机溶剂等对提取方法进行了优化,由于两种待测物的极性差异较大,液液萃取所得的提取回收率均低于50%,所以最终采用乙腈沉淀法进行提取。另外,为了尽量降低血浆中内源代谢物对于待测物的干扰,在进样前将样品用去磷脂板过滤,以消除磷脂类小分子对于待测物的离子抑制效应(图1)。另外,为了获得最佳的提取效果,分别采用所用血浆体积1倍,2倍,3倍,5倍,7.5倍的乙腈对待测物提取,结果表明采用7.5倍乙腈时提取效果最好(图2)。
在生物样本的测定中,选择合适的内标可以提高质谱方法的准确度和精密度。理想的内标与样品中的被分析物应有相似的理化性质和响应特征。本研究对比了人参皂苷rg1和地高辛作为内标的效果(图3),其中,地高辛与黄芪甲苷-iv有较为相近的保留时间和响应特征,可以作为定量方法中的内标来提高本方法的准确度和精密度。
3.2lc-ms条件的优化
首先采用蠕动泵进样的方式对黄芪甲苷-iv、环黄芪醇及其内标地高辛的质谱检测条件分别采用esi与apci两种离子源的正负离子模式进行了考察和优化。结果发现,待测物的离子化效率在esi条件下优于apci,且正离子模式较负离子模式响应高。在正离子模式下,待测物及内标的主要分子离子为[m+na]+,其质荷比分别为m/z807.4501(ast),m/z513.3550(cst),m/z803.4197(地高辛)。随后对鞘气、辅助气、反吹气等质谱参数进行了进一步优化,以获得待测物定量离子的最大质谱响应强度。
sim和平行反应监测(parallelreactionmonitoring,prm)是两种在orbitrap质谱上最常用的定量方法。本研究分别采用prm和sim的离子监测方法对待测物定量,发现两种待测物用sim方法定量的灵敏度和线性优于prm方法,尤其是cst因具有皂苷结构且没有支链,难于电离,并且由于cst的强亲脂性和与磷脂类分子相近的相对分子质量,在检测过程中容易受到基质效应的干扰,故采用sim方法对两种待测物定量,监测离子的质荷比为黄芪甲苷-ivm/z807.4501,环黄芪醇m/z513.3550,地高辛m/z803.4197。
此外,在sim方法检测时,质谱分辨率对于方法的灵敏度和专属性会有较大的影响。随着质谱分辨率从15,000增加到120,000,未能通过液相色谱柱分离的共流出物能够逐渐通过质谱有效分离(图5),使专属性显著提高。这种现象在cst的检测中尤其明显(图5b)。但是随着ms分辨率的进一步增加,质谱扫描速度呈逐渐降低趋势(图6),反而会影响检测灵敏度。当质谱分辨率为240,000时,可以看到ast和cst的信号强度显著降低,在500,000的分辨率下几乎检测不到两种待测物的信号。因此,最终选择120,000为m/z200时的分辨率。最后,将喷雾电压,蒸发器温度,鞘气,辅助气体,扫气和隔离窗口宽度等其它质谱参数也进行了优化,以最大限度地提高待测物的响应强度。
为了能在尽可能短的时间内对两种待测物定量以及获得较好的分离效果和峰型,对色谱条件进行了优化。首先对于不同的流动相进行选择,包括不同比例的甲醇/水,乙腈/水和甲醇/乙腈/水,结果发现使用均以甲醇/乙腈(1:1,v/v)和水作为流动相时ast和cst均有更好的响应。此外,在流动相中加入0.1%的甲酸可以增加色谱峰的对称性,有助于ast和cst的电离效率。随后测试了一系列不同长度和孔径的色谱柱。最终选择了zorbaxextend-c18rrhd(50×2.1mm,1.8μm)色谱柱,使用该色谱柱可以使分析时间最短并获得最佳的分离效果。经过条件优化后,梯度洗脱的总时间为3min,流速为0.5ml/min,ast、cst和内标的保留时间分别为1.93min,2.13min,1.66min。
3.3方法学验证
方法的专属性处理后的空白血浆样品,空白加标样品和给药动物的血浆样品的提取离子流色谱图见图7。结果表明小鼠血浆中的内源性物质对于黄芪甲苷-iv和环黄芪醇的测定无明显干扰。
线性范围与定量下限线性考察结果表明,本研究所建立的方法在1-200ng/ml的范围内线性关系良好,ast与cst的线性回归方程分别为y=0.0129x-0.0114(r2=0.9976)和y=0.0375x+0.0467(r2=0.9983)。黄芪甲苷-iv定量下限(lloq)1ng/ml和检测限(lod)为0.005ng/ml,环黄芪醇的定量下限为1ng/ml,检测限为0.016ng/ml。
准确度与精密度该方法的准确度和精密度数据见表1,其中,日内、日间精密度rsd均低于8.6%,准确度相对偏差re在8.8%以内,表明该方法准确度高,可靠性强。
表1.黄芪甲苷-iv(ast),环黄芪醇(cst)的准确度与精密度
a:平均实测浓度±标准偏差(sd)
b:sd/平均实测浓度×100
c:(实测-理论浓度)/理论浓度×100
提取回收率和基质效应两种待测物和内标的提取回收率和基质效应如表2所示。黄芪甲苷-iv、环黄芪醇和内标地高辛在1.5,15,150ng/ml三个浓度下的提取回收率均高于75%,基质效应rsd值均<15%。表明该方法重复性好。
稳定性待测物和内标在所有评估的储存条件下都是稳定的,待测物实测浓度均在理论浓度的±11.5%内。将加入标准溶液的血浆样品从10μg/ml稀释到1.5,15或150ng/ml时,发现三个浓度的实测浓度的re均低于±15%,表明当样品中待测物浓度超出该方法的线性范围时方法准确度不会受到显著影响。
表2.黄芪甲苷-iv(ast)、环黄芪醇(cst)和地高辛(digoxin)在小鼠血浆中的提取回收率和基质效应(n=18)
3.4药物代谢动力学
将建立的uplc-hrms方法应用于给药30mg/kgast后小鼠血浆样品中ast及其主要代谢物cst的定量。ast和cst的血药浓度-时间曲线如图8所示。灌胃2小时后,ast在血浆中的浓度开始升高,4小时后达到血药浓度最大值(cmax)。与最初的灌胃剂量(每只小鼠约1.2mg)相比,ast的cmax(59.2ng/ml)表明其在小鼠体内生物利用度低。给药后3小时cst血药浓度开始升高,tmax为8h,cmax为474.9ng/ml。ast和cst的药物消除半衰期(t1/2)分别为1.85和0.32h。在连续给药7天后没有观察到显著的药物累积。cst的曲线下面积(2299.9h·ng/ml)远大于ast(289.8h·ng/ml),表明ast有可能是通过在体内水解为cst发挥其药效作用,具体的药物作用机制有待于进一步研究。
4结论
本研究建立了在小鼠血浆中同时定量黄芪甲苷-iv和环黄芪醇的快速、灵敏、专属性强的hplc-hrms方法。该定量方法仅需20μl血浆样品,定量下限可达到1ng/ml,分析总时间为3min;采用地高辛作为内标,提高了分析方法的准确度和精密度;提取过程中采取乙腈沉淀法与固相提取相结合的提取方法,处理方法简便,提高了提取效率;色谱分离采用c18色谱柱,提高了色谱分离效率,缩短了分析时间,可实现高通量分离分析;离子监测模式采用选择离子监测法,提高了方法的灵敏度。本方法经过系统的方法学验证,准确度高,日内日间精密度良好,在1-200ng/ml的范围内线性关系良好,且可以应用于实际样本的测定中。因此,本研究建立的lc-ms定量方法适用于小鼠血浆中黄芪甲苷-iv和环黄芪醇的同时定量分析。
- 还没有人留言评论。精彩留言会获得点赞!