一种测定蒽醌类成分含量的方法与流程
本发明涉及一种蒽醌类成分的微量提取检测方法。
(二)
背景技术:
决明子是豆科植物决明(cassiaobtusifolial.)或小决明(cassiatoral.)的干燥成熟种子。决明子具有清热明目、润肠通便的功效,用于治疗目赤涩痛、羞明多泪、头痛眩晕、目暗不明、大便秘结。现代药理研究表明,决明子具有较好的保肝、降血脂、降血压、抗氧化、抗衰老、改善肾功能、抑菌等作用,是国家卫生部门公布的药食同源的中药品种之一。决明子中含有多种成分,主要含蒽醌类成分,包括橙黄决明素(aurantio-obtusin),黄决明素(chrysoobtusin),决明蒽醌(obtusifolin),决明素(obtusin),芦荟大黄素(aloe-emodin),大黄酸(rhein),大黄素(emodin),大黄酚(chrysophanol),大黄素甲醚(physcion)等。中国药典2015版将大黄酚和橙黄决明素作为指标性成分控制决明子药材及相关制剂的质量。常用的提取决明子中有效成分的方法,如加热回流提取法、超声提取法、微波萃取法(mae)等,普遍存在有机溶剂用量大、环境污染大、耗时长、操作繁琐、提取效率低等缺点。
基质固相分散萃取(mspd)是一种建立在固相萃取基础上的样品前处理新技术,具有溶剂用量少、对环境污染少、萃取时间短、操作简洁、提取效率高等优点,近年来逐渐成为国内外的热门研究,并已经广泛地被应用于天然产物分析检测。目前,在基质固相分散萃取技术中,主要选用硅胶、氧化铝、c18、弗罗里硅土、活性炭等作为吸附剂。
螃蟹作为消费量最高的十大海产品之一,然而,只有一小部分的螃蟹作为食物被送上了餐桌,其余的通常被认为是垃圾并被随意丢弃,造成了蟹壳资源的浪费和由于其低生物降解而导致的环境恶化。因此,开发蟹壳作为生物吸附剂也是非常有意义的。蟹壳由壳聚糖,碳酸钙和一些蛋白质组成。其中,壳聚糖在其聚合链上具有游离的氨基,显示出较高的金属结合力,此外,由于其低成本、高机械强度、刚性结构、耐受剧烈条件的能力和高生物相容性等优点,被应用于吸附重金属和其他物质如磷酸盐和染料。由于蟹壳粉安全无毒,且具有良好的吸附性能,因此作为吸附剂用于基质固相分散。
(三)
技术实现要素:
本发明的目的在于解决常规方法存在的样品用量大、耗时长、有毒有机溶剂用量大的问题,建立一种更高效、更绿色环保的提取及检测决明子中蒽醌类成分的方法。
本发明要点在于将蟹壳粉作为基质固相分散萃取的吸附剂与决明子粉末中的目标分子吸附结合,从而使得目标分子从决明子粉末中分离出来,然后使用绿色环保的离子液体作为洗脱剂,得到含有目标分子的洗脱液。洗脱液采用hplc进行分离检测。相比药典提取方法,本发明的有机溶剂使用量大大减少,安全,经济,环保。
本发明的技术方案如下:
一种测定蒽醌类成分含量的方法,所述蒽醌类成分为橙黄决明素、黄决明素、决明蒽醌、大黄素,所述方法包括以下步骤:
(1)样品分析
将待测样品粉末与吸附剂混合,研磨均匀后加入洗脱剂,再用旋涡混合器进行涡旋混合,接着用孔径0.22μm滤膜过滤,取滤液离心,收集上清液即为提取液,将所得提取液进行高效液相色谱分析,得到样品谱图;
所述待测样品粉末与吸附剂的质量比为1:0.5~2,优选1:0.5;
所述研磨的时间为1~4min,优选3min;
所述涡旋的时间为1~4min,优选3min;
所述离心的速率为13000rpm,时间为6min;
所述待测样品例如为决明子;
所述吸附剂为蟹壳粉末;
所述洗脱剂为150~300mm(优选250mm)离子液体的水溶液,所述离子液体选自1-丁基-3-甲基咪唑六氟磷酸盐([bmim]pf6)、氯化1-十二烷基-3-甲基咪唑([domim]cl)、1-十二烷基-3-甲基咪唑硫酸氢盐([domim]hso4)或1-十二烷基-3-甲基咪唑硝酸盐([domim]no3),优选1-十二烷基-3-甲基咪唑硫酸氢盐([domim]hso4);
所述高效液相色谱分析的条件为:色谱柱:c18柱,4.6mm×250mm,5μm;流动相:乙腈/水体积比95:5的混合液、乙腈/水体积比5:95的混合液,所述混合液中含有体积分数0.1%的甲酸;流速:0.8~1.2ml/min,优选1ml/min;柱温:20~30℃,优选25℃;紫外检测波长:440nm;
(2)标准曲线建立
称取橙黄决明素、黄决明素、决明蒽醌、大黄素的标准物质,以甲醇为溶剂配制成混合标准溶液,将所得混合标准溶液在步骤(1)中的高效液相色谱分析条件下进行检测,得到标准物质谱图,以混合标准溶液中各标准物质的进样量为横坐标,标准物质谱图中相应标准物质的峰面积为纵坐标,分别绘制橙黄决明素、黄决明素、决明蒽醌、大黄素的标准曲线;
(3)获取检测结果
将步骤(1)所得样品谱图与步骤(2)所得标准物质谱图进行对照,获得样品中蒽醌类成分的定性结果;
将步骤(1)所得样品谱图中各蒽醌类成分的峰面积值分别代入步骤(2)所得相应的标准曲线中,计算获得样品中蒽醌类成分的含量。
本发明中,所述蟹壳粉末按如下方法制备:
将蟹壳用自来水冲洗,去除碎屑和粘液,再用纯净水洗涤,然后置于60~70℃的烘箱中干燥至恒重,将干燥好的蟹壳粉碎并过100目筛,接着在90~100℃下用20~25wt%的naoh水溶液对过筛蟹壳粉末进行1h的碱处理,以除去多余的蛋白质,之后用纯净水洗涤,并在60~70℃的电炉中干燥12h,最后再次粉碎,过200目筛,即得作为吸附剂的蟹壳粉末。
与现有技术相比,本发明的有益效果在于:
本发明采用基质固相分散与涡旋相结合的技术来提取决明子中的有效成分,并使用绿色的离子液体水溶液代替传统的有机溶剂作为洗脱剂,集提取与分离于一体,具有操作简单方便、提取分离时间短、提取效率高、安全环保、洗脱剂与样品用量少等优点。
目前,蟹壳粉主要集中使用在污水中重金属、磷酸盐和染料的去除,本发明首次将蟹壳粉作为吸附剂用于中药决明子中复杂基质的提取分离,具有低成本、安全无毒、高机械强度、刚性结构、耐受剧烈条件的能力、高生物相容性等优点。
(四)附图说明
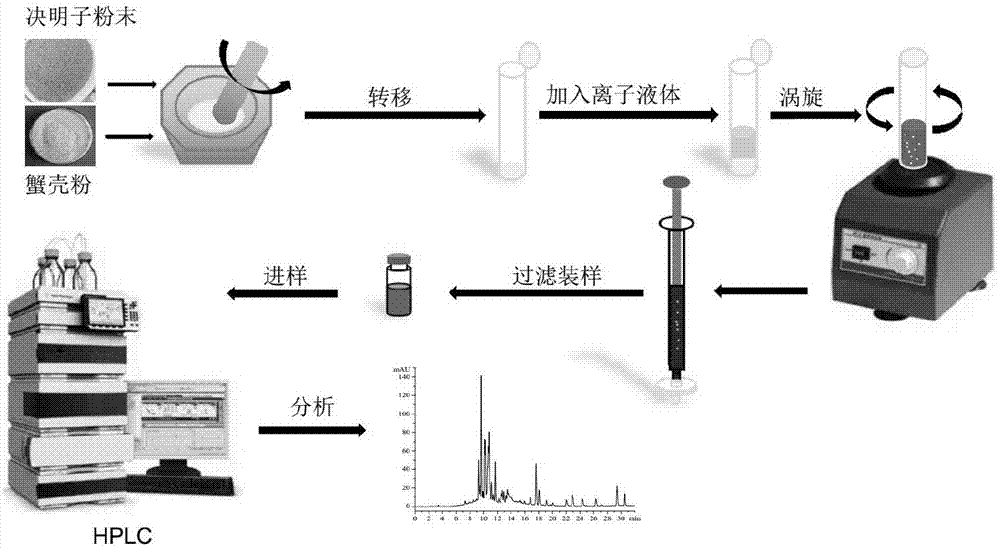
图1为本发明基质固相分散萃取方法的工艺流程图;
图2为蟹壳粉的表征:(a)蟹壳粉低倍(2000倍)放大的扫描电镜图像;(b)蟹壳粉高倍(50000倍)放大的扫描电镜图像;(c)蟹壳粉的ft-ir光谱;
图3为考察基质固相分散萃取工艺中的萃取效果柱状图;图中,1橙黄决明素;2黄决明素;3决明蒽醌;4大黄素;(a)样品与吸附剂的质量比;(b)研磨时间;(c)涡旋时间;
图4为考察洗脱剂对四种蒽醌提取效果的折线图;图中,1橙黄决明素;2黄决明素;3决明蒽醌;4大黄素;(a)离子液体的类型;(b)离子液体的浓度。
(五)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此。
本发明实施例所用决明子粉末根据中国药典2015年版制备。
实施例1蟹壳粉的制备
蟹壳用自来水冲洗,去除无用的其他碎屑和粘液。再将它们用纯净水洗涤,放在在60℃的烘箱中干燥至恒重。将干燥好的蟹壳粉碎成粉末并过100目筛。然后,在100℃下用20%(wt)naoh对过筛蟹壳粉末进行约1小时的碱处理,除去多余的蛋白质。用纯净水洗涤粉末,并在60℃的电炉中干燥约12小时。将经处理的粉末再次粉碎,过200目筛,即得作为吸附剂的蟹壳粉末,供样品前处理使用。
实施例2样品与吸附剂的质量比的选择
平行称取4份决明子粉末20mg分别加入到4组研钵中,并称取不同质量(0mg、10mg、20mg、40mg)的蟹壳粉,分别加入4组研钵中与决明子粉末研磨3min。取4支10ml离心管,分别将研磨好的固体混合物转移至离心管中。分别加入1ml的[domim]cl水溶液(250mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液即为决明子提取液,装样,用高效液相色谱仪进行采样分析,分析条件如下:
本实验采用的仪器为安捷伦高效液相色谱系统(agilent1260infinitylc,agilenttechnologies,santaclara,ca,usa)配备真空抽气泵,二元流动相系统,恒温自动进样器,恒温柱温箱。色谱柱inersustainc18column(4.6×250mm,5μm),柱温25℃。流动相:乙腈/水体积比95:5的混合液(流动相a)、乙腈/水体积比5:95的混合液(流动相b),所述混合液中含有体积分数0.1%的甲酸。梯度洗脱:如表1所示,平衡时间5min。进样量:20μl,流速:1ml/min,检测波长:440nm。
表1梯度洗脱过程
不同样品与吸附剂的质量比的萃取效果柱状图如图3中a所示,结果表明,当样品与吸附剂的质量比从1:0升到2:1时,萃取效果增强,且当样品与吸附剂的质量比为2:1时,对于所有研究的蒽醌吸附效果是最佳的。然而,当质量比提高到1:1或1:2时,萃取效果缓慢减弱,这可能是因为过多的吸附剂会使活性成分难以解吸,从而降低了萃取效果。
实施例3研磨时间的选择
平行称取4份决明子粉末20mg和4份10mg蟹壳粉分别加入到4组研钵中,分别研磨不同的时间(1min、2min、3min、4min)。取4支10ml离心管,分别将研磨好的固体混合物转移至离心管中。分别加入1ml的[domim]cl水溶液(250mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液即为决明子提取液,装样,用高效液相色谱仪进行采样分析(分析条件同实施例2)。
不同研磨时间的萃取效果柱状图如图3中b所示,结果表明,随着研磨时间的增加萃取效果增强,增至最优值3min后,当研磨时间延长至4min时,萃取效果反而下降。这可能是由于研磨时间过长导致活性成分被过度提取而被挤压到吸附剂中致密的孔内,增加了洗脱的难度,从而使萃取效果下降。
实施例4涡旋时间的选择
平行称取4份决明子粉末20mg和4份10mg蟹壳粉分别加入到4组研钵中,研磨3min。取4支10ml离心管,分别将研磨好的固体混合物转移至离心管中。分别加入1ml的[domim]cl水溶液(250mm),分别用旋涡混合器进行涡旋混合不同的时间(1min、2min、3min、4min),并过0.22μm的有机滤膜,13000rpm离心6min,取上清液即为决明子提取液,装样,用高效液相色谱仪进行采样分析(分析条件同实施例2)。
不同涡旋时间的萃取效果柱状图如图3中c所示,结果表明,随着涡旋时间的增加萃取效果增强,增至最优值3min后,当涡旋时间延长至4min时,萃取效果并没有显著的增加,说明涡旋3min足够把蟹壳粉上的活性成分洗脱下来。
实施例5离子液体种类的选择
平行称取4份决明子粉末20mg和4份10mg蟹壳粉分别加入到4组研钵中,研磨3min。取4支10ml离心管,分别将研磨好的固体混合物转移至离心管中。分别加入1ml的[domim]cl水溶液(250mm)、[domim]hso4水溶液(250mm)、[domim]no3水溶液(250mm)、[bmim]pf6水溶液(250mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液即为决明子提取液,装样,用高效液相色谱仪进行采样分析(分析条件同实施例2)。
不同离子液体种类对四种蒽醌提取效果的折线图如图4中a所示,[bmim]pf6对橙黄决明素具有最佳的洗脱效率,但是对黄决明素,决明蒽醌和大黄素的提取效率不是很好,这可能是由于[bmim]pf6和三种活性成分之间的极性不同。[domim]cl,[domim]hso4和[domim]no3虽然具有相同的长链阳离子,但对四种活性成分的洗脱效率不同,其中[domim]hso4和[domim]no3的提取效率比[domim]cl好,这可能是因为阴离子cl-和分析物之间具有较弱的静电。综合考虑,用[domim]hso4作为洗脱溶剂可以获得更好的萃取效率。
实施例6离子液体浓度的选择
平行称取4份决明子粉末20mg和4份10mg蟹壳粉分别加入到4组研钵中,研磨3min。取4支10ml离心管,分别将研磨好的固体混合物转移至离心管中。分别加入不同浓度的1ml的[domim]hso4水溶液(150mm、200mm、250mm、300mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液即为决明子提取液,装样,用高效液相色谱仪进行采样分析(分析条件同实施例2)。
不同离子液体浓度对四种蒽醌提取效果的折线图如图4中b所示,结果表明,随着[domim]hso4的增加洗脱效果增强,增至最优值250mm后,当[domim]hso4浓度增加至300mm时,洗脱效果并没有显著的增加,这可能是由于溶液粘度增加,导致难以将蒽醌从决明子中转移到[domim]hso4水溶液中。
实施例75种不同批次决明子中蒽醌类成分的定量
1、分别称取5种不同批次决明子粉末各20mg和5份10mg蟹壳粉分别加入到5组研钵中,研磨3min。取5支10ml离心管,分别将研磨好的固体混合物转移至离心管中。加入1ml的[domim]hso4水溶液(250mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液装样,用高效液相色谱仪进行采样分析(分析条件同实施例2)。
5种不同批次决明子中蒽醌类成分的定量如表2所示:
表25种不同批次决明子中四种活性成分的测定结果(mg/g)
a生决明子
b炒决明子
2、标准曲线的制定:
分别精密称取橙黄决明素、黄决明素、决明蒽醌和大黄素的对照品,分别用甲醇溶解配成浓度为1.03mg/ml、0.5mg/ml、0.52mg/ml和0.22mg/ml的对照品溶液。将制得的对照品溶液配制成含橙黄决明素123.6μg/ml、黄决明素40.0μg/ml、决明蒽醌41.6μg/ml、大黄素13.2μg/ml的混合对照品溶液。分别吸取0.5μl、1μl、2μl、5μl、10μl、15μl和20μl的混合对照品溶液注入液相色谱仪进行分析,测得峰面积。以进样量为横坐标,峰面积为纵坐标,绘制标准曲线。
橙黄决明素、黄决明素、决明蒽醌、大黄素的标准曲线、线性相关系数、线性范围和检测限如下表3:
表3各成分标准曲线方程、线性相关系数、线性范围和检测限
3、重复性考察
称取决明子粉末20mg和10mg蟹壳粉于研钵中,研磨3min。将研磨好的固体混合物转移至10ml的离心管中。加入1ml的[domim]hso4水溶液(250mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液装样。按照上述方法平行制样6个,在上述色谱条件下考察其重复性,结果见表4。
4、精密度考察
在上述色谱条件下,精密吸取混合对照品溶液同一天内重复进样6次,为日内精密度。在上述色谱条件下,精密吸取标准品溶液,连续3天每天进样2次,为日间精密度。结果见表4。
5、稳定性考察
称取决明子粉末20mg和10mg蟹壳粉于研钵中,研磨3min。将研磨好的固体混合物转移至10ml的离心管中。加入1ml的[domim]hso4水溶液(250mm),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液装样,在上述色谱条件下,分别于2、4、8、16、24h测定峰面积,考察其稳定性,结果见表4。
表4重复性、精密度、稳定性实验结果
表中数据显示,本发明方法重复性好、稳定性好,仪器的精密度好。
6、加样回收考察
称取决明子粉末10mg和10mg蟹壳粉于研钵中,研磨3min。将研磨好的固体混合物转移至10ml的离心管中。加入1ml的[domim]hso4水溶液(250mm)(含9.11μg/ml橙黄决明素、4.20μg/ml黄决明素、2.93μg/ml决明蒽醌、0.77μg/ml大黄素),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液装样,在上述色谱条件下进行分析。
称取决明子粉末10mg和10mg蟹壳粉于研钵中,研磨3min。将研磨好的固体混合物转移至10ml的离心管中。加入1ml的[domim]hso4水溶液(250mm)(含18.22μg/ml橙黄决明素、8.40μg/ml黄决明素、5.86μg/ml决明蒽醌、1.54μg/ml大黄素),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液装样,在上述色谱条件下进行分析。
称取决明子粉末10mg和10mg蟹壳粉于研钵中,研磨3min。将研磨好的固体混合物转移至10ml的离心管中。加入1ml的[domim]hso4水溶液(250mm)(含27.33μg/ml橙黄决明素、12.40μg/ml黄决明素、8.79μg/ml决明蒽醌、2.31μg/ml大黄素),用旋涡混合器进行涡旋混合3min,并过0.22μm的有机滤膜,13000rpm离心6min,取上清液装样,在上述色谱条件下进行分析。
加样回收率实验结果如下表5:
表5加样回收率实验
表中数据显示,本发明方法回收率高。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!