一种基于UPLC-QTRAP技术快速同时检测样本中23种人参皂苷的方法与流程
本发明属于分析检测技术领域,具体涉及一种基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法。
背景技术:
传统的中药分析方法主要基于一种药材分离纯化,将得到的纯化合物进行四大光谱分析,且常常因成分含量低不能纯化而难以鉴别。除了系统分离外,液相色谱-质谱法可能鉴定出三七中可能存在的多种皂苷类成分,其中以原人参二醇型三萜皂苷和原人参三醇型三萜皂苷最多。然而,以往研究使用的液相色谱-质谱仪多为低分辨率,所提供的裂解信息有限,而三七、人参中的皂苷存在大量的同分异构体,它们具有相同的相对分子质量,相似的特征峰,甚至相似的碎片信息,这给分析带来了很大的困难,同分异构体信号之间容易相互干扰,造成无法同时检测多种皂苷。
因此,如何提供一种快速识别人参皂苷化合物对照品,同时快速准确检测更多种人参皂苷是本领域有待解决的技术问题。
技术实现要素:
本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
鉴于上述的技术缺陷,提出了本发明。
因此,作为本发明其中一个方面,本发明克服现有技术中存在的不足,提供一种基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法。
为解决上述技术问题,本发明提供了如下技术方案:一种基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法,其包括,
人参样本的预处理:向人参样本中加入70%的甲醇水溶液,超声提取,取上清;
检测:调整液相色谱参数,色谱柱为thermoc18柱,i.d.2.1×100mm,2.6μm流动相a为水,流动相b为乙腈,柱温为45℃;流速为0.35ml/min;并调整质谱参数。
作为本发明所述的基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法的一种优选方案:所述调整液相色谱参数,流动相梯度条件为:
0min:95%流动相a、5%流动相b;
10min:5%流动相a、95%流动相b;
11min:5%流动相a、95%流动相b;
11.1min:95%流动相a、5%流动相b;
14min:95%流动相a、5%流动相b。
作为本发明所述的基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法的一种优选方案:所述调整质谱参数为:
时间段3.5-4.5min内:人参皂苷rd定量离子对931.53→475.4,碰撞能量-62v,驻留时间50ms;人参皂苷re定量离子对945.55→783.5,碰撞能量-40v,驻留时间50ms;人参皂苷rg1定量离子对799.49→637.4,碰撞能量-30v,驻留时间50ms;
时间段4.50-6min内:拟人参皂苷f1l定量离子对801.49→439.6,碰撞能量30v,驻留时间50ms;三七皂苷r2定量离子对769.48→475.3,碰撞能量-50v,驻留时间100ms;拟人参皂苷-rt5定量离子对655.43→457.2,碰撞能量-20v,驻留时间50ms;人参皂苷ro定量离子对955.50→793.4,碰撞能量-20v,驻留时间50ms;人参皂苷rg2定量离子对783.5→475.4,碰撞能量-50v,驻留时间50ms;人参皂苷rh1定量离子对637.44→475.4碰撞能量-50v,驻留时间50ms;人参皂苷rb1定量离子对1107.60→945.60,碰撞能量-60v,驻留时间50ms;原人参二醇定量离子对459.39→401.1,碰撞能量-20v,驻留时间50ms;人参皂苷rc定量离子对1077.59→945.2,碰撞能量-60v,驻留时间50ms;人参皂苷rd定量离子对945.55→783.40,碰撞能量-50v,驻留时间50ms;
时间段6-7.3min内:人参皂苷rb2定量离子对1077.59→945.6,碰撞能量-60v,驻留时间50ms;20(s)-人参皂苷rb3定量离子对1077.59→783.60,碰撞能量-58v,驻留时间100ms;人参皂苷-f1定量离子对637.44→475.1,碰撞能量-30v,驻留时间100ms;人参皂苷f2定量离子对783.50→621.5,碰撞能量-30v,驻留时间100ms;
时间段7.3-8min内:20(s)-人参皂苷rg3定量离子对783.5→459.5,碰撞能量-56v,驻留时间50ms;20(r)-人参皂苷rg3定量离子对783.5→459.5,碰撞能量-59v,驻留时间50ms;
时间段8-10min内:原人参三醇定量离子对475.39→391.4,碰撞能量-40v,驻留时间50ms;人参皂苷rk1定量离子对765.49→603.2,碰撞能量-40v,驻留时间50ms;人参皂苷k定量离子对621.44→459.3,碰撞能量-30v,驻留时间50ms。
作为本发明所述的基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法的一种优选方案:还包括,将2-氯苯丙氨酸代替溶剂作为内标,在检测过程中,通过2-氯苯丙氨酸峰面积和保留时间反映实际样本的采集情况;所述2-氯苯丙氨酸浓度为1μg/ml。
作为本发明所述的基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法的一种优选方案:还包括,使用4,4’-亚甲基双(2-氯苯胺)、对茴香胺、l-酪氨酸甲酯、3-氯苯胺、2,4-二甲基喹啉、磺胺吡啶、阿特拉津、磺胺多辛、dl-亮氨酸、n-苯甲酰基-l-酪氨酸乙酯、6-苯基-2-硫脲嘧啶、n-(邻甲苯酰)甘氨酸、2-甲基-5-硝基咪唑-1-乙醇、甘草次酸、黄烷酮、ε-己内酯、2-氨基吡啶的混合标样作为通用质控品,每次检测至少进两针通用质控品,根据谱图重叠情况分析检测仪器的状态。
作为本发明所述的基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法的一种优选方案:以质量比计,所述4,4’-亚甲基双(2-氯苯胺):对茴香胺:l-酪氨酸甲酯:3-氯苯胺:2,4-二甲基喹啉:磺胺吡啶:阿特拉津:磺胺多辛:dl-亮氨酸:n-苯甲酰基-l-酪氨酸乙酯:6-苯基-2-硫脲嘧啶:n-(邻甲苯酰)甘氨酸:2-甲基-5-硝基咪唑-1-乙醇:甘草次酸:黄烷酮:ε-己内酯:2-氨基吡啶=1:0.5:0.5:2:1:0.4:0.1:0.1:0.2:0.5:0.5:1:1:0.1:0.5:2:0.2。
作为本发明所述的基于uplc-qtrap技术快速同时检测样本中23种人参皂苷的方法的一种优选方案:所述4,4’-亚甲基双(2-氯苯胺)的浓度为1μg/ml、对茴香胺的浓度为0.5μg/ml、l-酪氨酸甲酯的浓度为0.5μg/ml、3-氯苯胺的浓度为2μg/ml、2,4-二甲基喹啉的浓度为1μg/ml、磺胺吡啶的浓度为0.4μg/ml、阿特拉津的浓度为0.1μg/ml、磺胺多辛的浓度为0.1μg/ml、dl-亮氨酸的浓度为0.2μg/ml、n-苯甲酰基-l-酪氨酸乙酯的浓度为0.5μg/ml、6-苯基-2-硫脲嘧啶的浓度为0.5μg/ml、n-(邻甲苯酰)甘氨酸的浓度为1μg/ml、2-甲基-5-硝基咪唑-1-乙醇的浓度为1μg/ml、甘草次酸的浓度为0.1μg/ml、黄烷酮的浓度为0.5μg/ml、ε-己内酯的浓度为2μg/ml、2-氨基吡啶的浓度为0.2μg/ml。
本发明的有益效果:本发明通过控制液相色谱流动相梯度条件,并确定23种皂苷同时检测时的最佳质谱参数,并采用2-氯苯丙氨酸作为通用内标监控样本采集情况,对23种人参皂苷进行定量分析,实现了以快速、便捷、灵敏的同时测定人参中23种人参皂苷的含量,排除采集有问题的样本,并将17个混标作为通用质控品监控仪器波动状态,排除每次实验的混标容易存在本身响应不佳、信号相互干扰、重复性差等技术问题,大大简化了高通量检测中繁琐的操作和复杂的信号分析、数据处理的问题,并使得检测结果更加准确可靠。本发明实现了同时测定人参中23种人参皂苷,总分析时长11分钟,现有方法最多只能检测不超过19种人参皂苷,而且检测时间为120min,因此本发明扩展了现有方法的检测范围,显著提高了分析效率。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
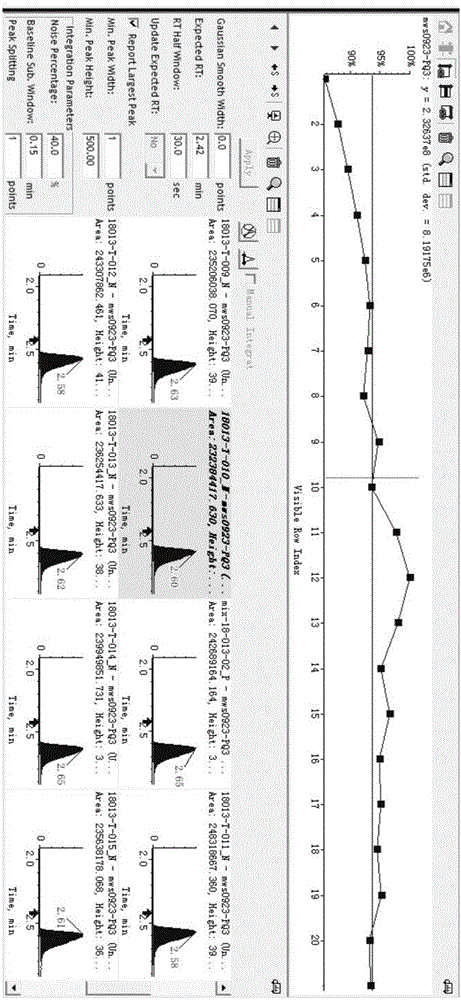
图1为本发明方法multiquant软件参数设置示范。
图2为本发明检测的样本内标异常的数据图。
图3为本发明检测的异常样本的出峰图。
图4为本发明重新采集后正常样本的出峰图。
图5为本发明质控品在液相色谱-质谱联用仪器上出峰情况图。
图6为本发明质控品在液相色谱-质谱联用仪器上的峰重叠情况图。
图7为在一次验证实验中,本发明混标质控品的出峰及峰重叠情况图。
图8为在一次验证实验中,已知样本的出峰及峰重叠情况图。
图9为在一次验证实验中,本发明混标质控品的出峰情况图。
图10为在一次验证实验中,已知样本的出峰及峰重叠情况图。
图11为在一次验证实验中,本发明混标质控品的出峰及峰重叠情况图。
图12为在一次验证实验中,已知样本的出峰及峰重叠情况图。
图13为本发明实施例1人参样本同时检测的23种人参皂苷的tic图。
图14为本发明实施例2人参样本同时检测的23种人参皂苷的tic图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
实施例1:
本发明采用超高效液相色谱-三重四级杆/线性离子阱串联质谱(uplc-qtrap)技术,质谱仪为absciexq-trap6500,分析软件为multiquant。
人参样本的预处理:
取待测人参样本适量,加入70%的甲醇水溶液,超声提取10min,取上清,过0.22um的滤膜,待分析;
确定超高效液相色谱-三重四级杆/线性离子阱串联质谱仪的运行参数:
液相相色谱运行条件:
色谱柱:thermoc18柱,i.d.2.1×100mm,2.6μm
流动相a:水(含0.04%乙酸)流动相b:乙腈(含0.04%乙酸)柱温:45℃;流速:0.35ml/min;进样量:2ul
流动相梯度条件:
质谱运行条件:
23种人参皂苷的质谱参数:
时间段3.5-4.5min内:人参皂苷rd定量离子对931.53→475.4,碰撞能量-62v,驻留时间50ms;人参皂苷re定量离子对945.55→783.5,碰撞能量-40v,驻留时间50ms;人参皂苷rg1定量离子对799.49→637.4,碰撞能量-30v,驻留时间50ms;
时间段4.50-6min内:拟人参皂苷f1l定量离子对801.49→439.6,碰撞能量30v,驻留时间50ms;三七皂苷r2定量离子对769.48→475.3,碰撞能量-50v,驻留时间100ms;拟人参皂苷-rt5定量离子对655.43→457.2,碰撞能量-20v,驻留时间50ms;人参皂苷ro定量离子对955.50→793.4,碰撞能量-20v,驻留时间50ms;人参皂苷rg2定量离子对783.5→475.4,碰撞能量-50v,驻留时间50ms;人参皂苷rh1定量离子对637.44→475.4碰撞能量-50v,驻留时间50ms;人参皂苷rb1定量离子对1107.60→945.60,碰撞能量-60v,驻留时间50ms;原人参二醇定量离子对459.39→401.1,碰撞能量-20v,驻留时间50ms;人参皂苷rc定量离子对1077.59→945.2,碰撞能量-60v,驻留时间50ms;人参皂苷rd定量离子对945.55→783.40,碰撞能量-50v,驻留时间50ms;
时间段6-7.3min内:人参皂苷rb2定量离子对1077.59→945.6,碰撞能量-60v,驻留时间50ms;20(s)-人参皂苷rb3定量离子对1077.59→783.60,碰撞能量-58v,驻留时间100ms;人参皂苷-f1定量离子对637.44→475.1,碰撞能量-30v,驻留时间100ms;人参皂苷f2定量离子对783.50→621.5,碰撞能量-30v,驻留时间100ms;
时间段7.3-8min内:20(s)-人参皂苷rg3定量离子对783.5→459.5,碰撞能量-56v,驻留时间50ms;20(r)-人参皂苷rg3定量离子对783.5→459.5,碰撞能量-59v,驻留时间50ms;
时间段8-10min内:原人参三醇定量离子对475.39→391.4,碰撞能量-40v,驻留时间50ms;人参皂苷rk1定量离子对765.49→603.2,碰撞能量-40v,驻留时间50ms;人参皂苷k定量离子对621.44→459.3,碰撞能量-30v,驻留时间50ms。
同时,本发明选用2-氯苯丙氨酸作为内标,配制成1μg/ml的内标提取液,用内标2-氯苯丙氨酸提取液代替纯溶剂对实验样本进行提取,保证每个样本中内标的浓度一致,上机采集数据,在数据采集过程中,通过multiquant软件参数设置,查看内标峰面积和保留时间,通过内标的采集情况,反映实际样本的采集情况,内标异常的个别样本,再打开对应的采集数据查看,数据异常的进行重采。
multiquant软件参数设置:删除除内标以外的其它物质;生成所有样本内标的结果表格;选中面积一列,点击createsametricplot,生成面积折线图,将折线图纵坐标改成百分比的形式,添加一条代表内标面积平均值横线,样本内标面积在平均值上下10%范围内,认为样本数据采集正常,超出10%再返回数据采集软件查看样本采集数据,异常样本重新提交采集;选中保留时间一列,点击createsametricplot,生成保留时间折线图,偏差超过0.1的返回数据采集软件查看样本采集数据,异常样本重新提交采集。
本发明从众多物质中经过反复筛选,选择2-氯苯丙氨酸作为内标,发现它在正负离子模式下都能够出峰且峰型好,并且不容易对待测物质产生干扰,采用本发明方法能够准确监测样本的采集情况,且不会发生遗漏,例如,当检测样本在加样过程中掺入气泡时,会造成该样本体积减少,造成出峰响应降低,使得最终该样本的定量不准确,现有技术方法无法监控到这一技术问题,采用本发明内标质控品后,当某一样本掺入气泡时,该样本中加入的内标的出峰响应也会降低,因此能够准确发现该采集情况不佳的样本。
而当选择其他物质作为内标,例如3-碘化酪氨酸、十五氟辛酸、三氯蔗糖、3-氯苯胺、十三烷酸、甘氨酸、苯甲酸、琥珀酸等时,由于其不稳定、或正负离子模式下影响差别太大、或样本物质易产生干扰、检测重复性不佳,因此不适于作为通用内标。
图1为本发明方法multiquant软件参数设置。图2为本发明内标异常的样本,图3为该样本的出峰图,从图2和图3可知,当内标发生异常时,说明样本采集异常,图4为将该样本重新采集进样后的出峰图,对比图3可知,图3样本采集时出峰响应明显下降,说明本发明选择2-氯苯丙氨酸作为内标能够准确反映样本采集情况。
为了能够同时监控检测时仪器的波动状态,本发明筛选17个混标组成通用质控品,本发明的通用质控品17个标样及其使用终浓度见表1:
表1
本发明通过反复测试,挑选分离度好、响应合适、稳定的化合物作为混标,17个标样出峰时间基本均匀分布在0~11分钟,相互对彼此出峰时间影响较小,响应在1*10^6~3*10^7之间,属于所用测试仪器最佳响应范围,17个标样化合物结构稳定,不易挥发、变质,两次采集之间各物质响应波动在20%以内,将数百个潜在的可能作为标准品的物质分别配制成1mg/ml的母液和1μg/ml的工作液,用工作液进行标准品的液质方法开发,确定检测条件,再用母液配制成一个混合标准品溶液,上机采集,并调整标准品的浓度,配成混标,上机采集数据,由于不同组合的标样之间可能会相互反应造成质控品不稳定、或各标样的检测峰之间容易相互干扰、或者响应不佳等,因此本发明经过反复组合、筛选,最终确定17个能够适用于各种不同类别物质检测的混标质控品,17个混标质控品色谱出峰图如图5所示,从图5可知17个标样检测峰相互分开,基本不互相影响,且响应良好,在所用仪器最适范围内,17个混标总离子流色谱图重叠图如图6所示。从图6可以看出两针混标质控品重叠很好,峰高一致,表明仪器响应稳定,保留时间一致,表明色谱分离情况稳定,仪器状态良好。本发明质控品能够作为通用质控品,适用于所有物质的检测质控品。
在数据采集过程中通过前后采集的数据总离子流色谱图重叠情况判断仪器波动情况,色谱图重叠较好说明仪器稳定,重叠不好则通过重叠情况判断仪器状态,例如保留时间重叠不上说明可能是柱压或柱温不稳定,峰高重叠不上说明可能是色谱柱堵塞或质谱污染等。
本发明混标质控品十分稳定,且配制简单,用量少,成本低。本发明经过针对不同种类物质的多次重复检测证明,采用本发明质控品能够精准的反应色谱仪或质谱仪的仪器状态(仪器是否稳定、柱压、柱温是否稳定、柱子是否有堵塞、是否有污染等)以及待测样本的状态,并能够根据谱图重叠情况成功分析出仪器不稳定的原因,判断准确度达到100%。
以不同状态的已知样本作为标准,来验证本发明的质控品对于不同样本状态和仪器状态的监控准确度,每个已知样本做一次重复进样,每一个混标质控品做一次重复进样,已知样本的出峰及重叠情况若与本发明混标质控品的出峰及重叠情况一致,则说明本发明混标质控品对于样本及仪器状态的情况反映准确。经过多次实验验证,发现本发明混标质控品能够100%反映样本采集情况及采样时仪器的运行状态,通过混标质控品的出峰及重叠情况,即可反映待测样本的采集情况。
如图7和图8所示,图7为在一次验证实验中,本发明混标质控品的出峰及峰重叠情况,如图7所示,两针混标质控品重叠很好,峰高一致,表明仪器响应稳定,保留时间一致,表明色谱分离情况稳定,仪器状态良好,如图8所示,在本次验证实验中,已知样本的出峰及重叠同样很好,说明本发明混标质控品能够精确反应仪器的正常状态。
如图9和图10所示,图9为在一次验证实验中,本发明混标质控品的出峰情况,从图9看出,质控品峰型不好、分离不好,且10min左右混标质控品未出峰,说明检测时色谱仪器存在问题。图10为本次验证实验中,已知样本的出峰及重叠情况,从图10看出,已知样本同样分离不好,并且8~10min响应变低,本发明质控品与已知样本响应一致,并且经过验证,色谱仪在检测时存在问题。说明本发明混标质控品能够精确反应仪器的异常状态。
如图11和图12所示,图11为在一次验证实验中,本发明混标质控品的出峰及峰重叠情况,如图11所示,两针混标质控品峰重叠不好,1~3min保留时间偏移,说明监测时仪器不稳定,图12为本次验证实验中,已知样本的出峰及重叠情况,从图12看出,已知样本同样峰重叠不好,1~3min样本保留时间同样存在偏移,并且经过验证,色谱仪在检测时柱压不稳,说明本发明混标质控品能够精确反应仪器的异常状态。
本发明在实验过程中不必单独配制质控品,特别是当大批量检测时,本发明通用质控品尤为方便,由于人参中的皂苷存在大量的同分异构体,它们具有相同的相对分子质量,相似的特征峰,甚至相似的碎片信息,这给分析带来了很大的困难,同分异构体信号之间容易相互干扰,因此如果按照传统方法采用人参样本混合样作为质控品,不易准确监控仪器状态,而本发明能够根据谱图重叠情况成功分析出仪器不稳定的原因,判断准确度达到100%。
图13为本发明实施例1人参样本同时检测的23种人参皂苷的tic图。本发明通过控制液相色谱流动相梯度条件,并确定23种皂苷同时检测时的最佳质谱参数,并采用2-氯苯丙氨酸作为通用内标监控样本采集情况,对23种人参皂苷进行定量分析,实现了以快速、便捷、灵敏的同时测定人参中23种人参皂苷的含量,排除采集有问题的样本,并将17个混标作为通用质控品监控仪器波动状态,排除每次实验的混标容易存在本身响应不佳、信号相互干扰、重复性差等技术问题,大大简化了高通量检测中繁琐的操作和复杂的信号分析、数据处理的问题,并使得检测结果更加准确可靠。
本发明实现了同时测定人参中23种人参皂苷,总分析时长11分钟,现有方法最多只能检测不超过19种人参皂苷,而且检测时间为120min,因此本发明扩展了现有方法的检测范围,显著提高了分析效率。
实施例2(对照例):
将实施例1中流动相梯度条件更改设置为:
流动相梯度条件:
其余条件与实施例1相同。实施例2人参样本同时检测的23种人参皂苷的tic图如图14所示,从图14可知,更改本发明液相色谱流动相梯度条件,造成无法同时检测出23种皂苷,无法区分鉴别出相近异构体的皂苷。
综上,本发明通过控制液相色谱流动相梯度条件,并确定23种皂苷同时检测时的最佳质谱参数,并采用2-氯苯丙氨酸作为通用内标监控样本采集情况,对23种人参皂苷进行定量分析,实现了以快速、便捷、灵敏的同时测定人参中23种人参皂苷的含量,排除采集有问题的样本,并将17个混标作为通用质控品监控仪器波动状态,排除每次实验的混标容易存在本身响应不佳、信号相互干扰、重复性差等技术问题,大大简化了高通量检测中繁琐的操作和复杂的信号分析、数据处理的问题,并使得检测结果更加准确可靠。
本发明实现了同时测定人参中23种人参皂苷,总分析时长11分钟,现有方法最多只能检测不超过19种人参皂苷,而且检测时间为120min,因此本发明扩展了现有方法的检测范围,显著提高了分析效率。
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!