颈腰康胶囊近红外定量校正模型的构建方法及颈腰康胶囊的检测方法与流程
本发明涉及药物检测领域,特别涉及颈腰康胶囊近红外定量校正模型的构建方法及颈腰康胶囊的检测方法。
背景技术:
颈腰康胶囊由制马钱子、伸筋草、香加皮、乳香(醋炒)、红花等10味中药材组成,具有舒筋通络,活血去瘀,消肿止痛作用。用于骨折瘀血肿胀疼痛,骨折恢复期,以及肾虚挟瘀所致痹痛(增生性脊柱炎,腰椎间盘脱出症)。疗效确切,市场认可度较高。
目前在药品检测现有技术中,常采用高效液相色谱法对样品进行单指标成分检测,测定其中药效成分的含量。样品前处理复杂、耗时、耗力、成本高,并且高效液相色谱法检测时间相对较长,检验工作效率降低。
技术实现要素:
有鉴于此,本发明提供一种颈腰康胶囊近红外定量校正模型的构建方法及颈腰康胶囊的检测方法。该检测方法达到多指标同时检测、样品处理简单、无需消耗试剂、快速分析、绿色环保等目的。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种颈腰康胶囊近红外定量校正模型的构建方法,取颈腰康胶囊的内容物,以士的宁、羟基红花黄色素a和水分作为关键质控指标,分别经高效液相色谱检测和近红外光谱检测,获得相应的高效液相色谱检测结果和近红外光谱结果,经偏最小二乘法和交叉-验证法获得颈腰康胶囊近红外定量校正模型。
近红外(nir)光谱技术是一种现代高新分析技术,主要包括近红外光谱仪、化学计量学软件和应用模型3个部分,可同时分析多种成分,无污染,样品不需特别预处理,不使用有毒、有害试剂,不对样品造成损伤,可实时分析和远距离测定,操作简单,分析成本低等优点,被称为“绿色分析技术”。
声光可调滤光器(aotf)为第五代近红外光谱仪,近红外分光技术发展,由滤光片分光技术、光栅扫描分光技术、傅利叶变换分光技术、二极管阵列分光技术、aotf声光可调式分光技术的逐渐演变与完善,aotf作为一种分光器件,特别适用于近红外光谱仪,整个光路的搭建全是静态的,光谱仪器的体积比较小,稳定性和可靠性比较好,整个设计一体化。
本发明收集60批次颈腰康胶囊成品,其中选取含量高低均匀样本55批样品,用于建立校正模型,其余5批为验证样品,用于对模型进行外部验证。采用hplc法分别测定颈腰康胶囊中士的宁、羟基红花黄色素a,采用烘干法检测水分指标含量,取内容物适量,采用aotf近红外光谱仪扫描相应光谱,应用unscrambler定量分析建立颈腰康胶囊近红外光谱与士的宁、羟基红花黄色素a及水分含量测定值的关联模型。
在本发明的一些具体实施方案中,士的宁采用高效液相色谱法测定,所述高效液相色谱的条件为:
填充剂为硅胶;流动相为正己烷-二氯甲烷-甲醇-浓氨试液(42.5:42.5:5.4:0.35),等度洗脱;检测器为紫外检测器,检测波长为210~300nm;流速为0.5~1.5ml·min-1;进样量为5~20μl;柱温20~40℃;优选的,检测波长为254nm;流速为1ml·min-1;进样量为5μl;柱温30℃;理论塔板数按士的宁峰计算应不低于6000。
在本发明的一些具体实施方案中,羟基红花黄色素a采用高效液相色谱法测定,所述高效液相色谱的条件为:填充剂为十八烷基硅烷键合硅胶;流动相为甲醇-乙腈-0.025mol/l磷酸二氢钾溶液(用磷酸调至ph=2.5)(15:5:80),等度洗脱;检测器为紫外检测器,检测波长为350~420nm;流速为0.5~1.5ml·min-1;进样量为5~20μl;柱温20~40℃;优选的,检测波长为403nm;流速为1ml·min-1;进样量为10μl;柱温30℃;理论塔板数按羟基红花黄色素a峰计算应不低于3000。
在本发明的一些具体实施方案中,所述高效液相色谱检测中士的宁对照品溶液的制备方法为:取士的宁对照品,加乙酸乙酯制成每1ml含50μg的溶液,即得;
所述高效液相色谱检测中羟基红花黄色素a对照品溶液的制备方法为:取羟基红花黄色素a对照品,加25%甲醇制成每1ml含60μg的溶液,即得。
在本发明的一些具体实施方案中,所述高效液相色谱检测中士的宁供试品溶液的制备方法为:取颈腰康胶囊的内容物,加入三氯甲烷与浓氨试液,过滤,收集滤液,蒸干,残渣用乙酸乙酯溶解,即得;
所述高效液相色谱检测中羟基红花黄色素a供试品溶液的制备方法为:取颈腰康胶囊的内容物,加入25%甲醇,加热回流,冷却,用25%甲醇补足减失的重量,过滤,收集滤液,即得。
在本发明的一些具体实施方案中,所述近红外光谱检测包括光谱采集、光谱预处理的步骤;
所述光谱采集为:取颈腰康胶囊的内容物,在25℃±5℃条件下,采用漫反射内置光源采集近红外光谱,以空气为参比背景,扫描次数为200~400,扫描光谱范围为1100~2300nm,波长增量1~2nm,装样量厚度为1~2cm。
在本发明的一些具体实施方案中,所述光谱预处理为:将所述光谱采集的原始光谱,通过反射比到吸光度(reflectancetoabsonrbance)的格式转化,再进行一阶微分9点平滑法(savitzky-golay)处理。
在本发明的一些具体实施方案中,所述获得贞芪扶正制剂近红外定量校正模型具体为:用unscrambler定量分析软件,采用偏最小二乘法、交叉-验证法,将预处理后光谱数据与样品hplc含量数据关联,建立各指标的校正模型。
在本发明的一些具体实施方案中,颈腰康胶囊士的宁nir模型中,内部验证直线方法为y=0.9731x+0.0184,外部验证直线方程为y=0.9614x+0.0266,其中x为化验值,y为预测值,内部验证决定系数r2=0.9865,外部验证决定系数r2=0.9778,rmsec=0.0116,rmsep=0.0149;
颈腰康胶囊的羟基红花黄色素anir模型中,内部验证直线方法为y=0.9547x+0.0330,外部验证直线方程为y=0.9317x+0.0500,内部验证决定系数r2=0.9771,外部验证决定系数r2=0.9638,rmsec=0.0192,rmsep=0.0241;
颈腰康胶囊水分nir模型中,内部验证直线方法为y=0.9750x+0.1464,外部验证直线方程为y=0.9469x+0.3104,内部验证决定系数r2=0.9874,外部验证决定系数r2=0.9750,rmsec=0.1290,rmsep=0.1813。
本发明提供了一种颈腰康胶囊的检测方法,按照本发明所述的构建方法获得颈腰康胶囊近红外定量校正模型,将颈腰康胶囊待测样品的近红外光谱数据导入所述颈腰康胶囊近红外定量校正模型,获得所述待测样品中士的宁、羟基红花黄色素a和水分的含量。
本发明利用aotf-nir技术,建立颈腰康胶囊成品中士的宁、羟基红花黄色素a及水分指标nir模型,无需繁琐的样品处理,可同时、快速预测成品中多个指标含量,预测结果较准确。本发明提供的方法,不仅缩短了测检测周期、减少了耗材使用、降低了检测成本,而且能够实现现场分析。为颈腰康胶囊质量控制提供快速、高效的检测方法。
本发明所建立的模型预测精度高,准确度高,满足实际生产中定量分析的要求;该方法与传统药典方法相比,在保证准确度的基础上,大大提高了分析效率,可以快速、高效地对产品质量进行控制,从而有利于保证产品质量的安全性、稳定性和有效性。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
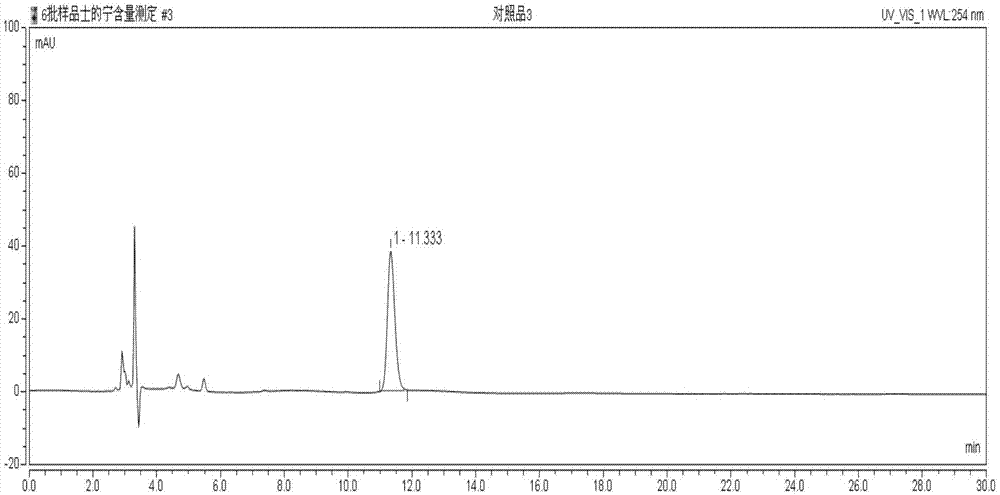
图1示士的宁对照品hplc图;
图2示供试品溶液hplc图;
图3示羟基红花黄色素a对照品hplc图;
图4示供试品溶液hplc图;
图5示颈腰康胶囊内容物nir光谱图;
图6示颈腰康胶囊内容物nir预处理光谱图;
图7示颈腰康胶囊-士的宁nir建模结果;
图8示颈腰康胶囊-羟基红花黄色素anir建模结果;
图9示颈腰康胶囊-水分nir建模结果;
对于建模结果:
粉色是不包括该点的模型对该点预测得出的验证值,蓝色是所有校正集样品建立的模型对校正集中每一个样品预测得出的校正值,两者都是交互验证过程对模型实际预测能力的模拟,但是粉色的验证值更接近外部预测,实际优化模型时,以外部决定系数为主。
具体实施方式
本发明公开了一种颈腰康胶囊近红外定量校正模型的构建方法及颈腰康胶囊的检测方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明提供了一种颈腰康胶囊nir快速检测方法,包括如下步骤:
(1)选取士的宁、羟基红花黄色素a及水分含量作为颈腰康胶囊质量控制关键指标;
(2)采集颈腰康胶囊内容物近红外光谱数据;
(3)选择近红外原始光谱预处理方法;
(4)建立颈腰康胶囊中质量控制关键指标的近红外定量校正模型:应用化学计量学软件将所得到的近红外光谱信息和参比方法所测得的标准值进行关联,建立近红外光谱和质量控制关键指标之间的定量校正模型;
(5)采集待测的颈腰康胶囊内容物的近红外光谱数据,导入步骤(4)所述定量校正模型,经模型预测得到待测样品中士的宁、羟基红花黄色素a含量和水分含量。
作为优选,所述步骤(2)通过以下步骤采集颈腰康胶囊近红外光谱数据,在室温条件下,称取颈腰康胶囊内容物粉末装入样品槽中,要求样品装样量厚度为1~2cm,以空气为背景,采用漫反射方式、比率模式下采集近红外光谱,光谱范围1100~2300nm;波长平均次数为200~400,波长增量1~2nm。
作为优选,所述步骤(3)通过以下步骤进行颈腰康胶囊近红外光谱预处理:将采集的nir光谱,通过反射比到吸光度(reflectancetoabsonrbance)的格式转化,再进行一阶微分9点平滑法(savitzky-golay)处理,即得预处理光谱。
作为优选,所述步骤(4)通过以下步骤建立颈腰康胶囊近红外校正模型:将预处理后的光谱数据与样品参比方法检测例如hplc含量数据关联,采用偏最小二乘法和交互验证方法,用unscrambler定量分析软件建立颈腰康胶囊中士的宁、羟基红花黄色素a及水分校正模型。
关键术语解释和技术缩略语:
aotf:声光可调滤光器
nir:近红外光谱技术
hplc:高效液相色谱法。
本发明提供的颈腰康胶囊近红外定量校正模型的构建方法及颈腰康胶囊的检测方法中所用原料、试剂及辅料均可由市场购得。
下面结合实施例,进一步阐述本发明:
实施例1颈腰康胶囊各指标含量测定
士的宁含量测定方法:按照国家药品标准
ws-10207(zd-0207)-2002-2012z检测,具体如下:
色谱条件及系统适应性试验以硅胶为填充剂;以正己烷-二氯甲烷-甲醇-浓氨试液(42.5:42.5:5.4:0.35)为流动相;检测波长为254nm,理论塔板数按士的宁峰计算应不低于6000。
对照品溶液的制备取士的宁对照品适量,精密成定,加乙酸乙酯制成每1ml含50μg的溶液,摇匀,即得。
供试品溶液的制备取装量差异项下的本品内容物,混匀,取约0.6g,精密成定,置具塞锥形瓶中,精密加入三氯甲烷50ml与浓氨试液1ml,密塞,称定重量,放置24小时,再称定重量,用三氯甲烷补足减失的重量,充分振摇,滤过,精密量取续滤液25ml,蒸干,残渣用乙酸乙酯溶解,转移至10ml量瓶中,加乙酸乙酯稀释至刻度,摇匀,即得。
测定法分别精密吸取对照品溶液与供试品溶液5μl,注入液相色谱仪,测定,即得。
羟基红花黄色素a含量测定方法:
色谱条件及系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇-乙腈-0.025mol/l磷酸二氢钾溶液(用磷酸调至ph=2.5)(15:5:80)为流动相;检测波长为403nm,理论塔板数按羟基红花黄色素a峰计算应不低于3000。
对照品溶液制备取羟基红花黄色素a对照品适量,精密称定,加25%甲醇制成每1ml含60μg的溶液,即得。
供试品溶液的制备取颈腰康胶囊内容物粉末约0.5g,精密称定,置具塞锥形瓶中,精密加入25%甲醇50ml,称定重量,加热回流60min,取出,放冷,再称定重量,用25%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定方法分别精密吸取对照品溶液与供试品溶液10μl,注入液相色谱仪,测定,即得。
水分含量测定方法:取供试品内容物,照水分测定法(通则0832第二法烘干法)测定。
对照品的hplc谱图见图1、图3;供试品的测定结果见图2、图4以及表1~表2。
表1颈腰康胶囊各指标含量测定结果
表2颈腰康胶囊水分测定结果
实施例2颈腰康胶囊内容物nir光谱采集
取不同批次样品,分别倒入样品槽中,用配套槽盖将样品摊平、压实,装样厚度1.5cm,在光谱范围1100~2300nm扫描,扫描间隔为1.0nm,扫描平均次数为300次。每个样品采集3次,即得到样品光谱图,如图5所示。
实施例3光谱预处理
在建立模型前,需要消除噪音和基线漂移等因素对光谱采集的影响。利用snap软件中的光谱处理程序,采用一阶微分9点平滑法(savitzky-golay)对原始光谱进行预处理,消除颜色差别等因素引起的光谱基线偏移和漂移。如图6所示。
实施例4建立pls数学模型
将各指标测定结果与经过预处理光谱进行关联,采用unscrambler定量分析软件,以偏最小二乘法(pls)和交叉-验证法(cross-validation)建立各指标校正模型。
采用光谱影响值leverage和化学值误差residual等统计量检验,剔除nir光谱与各指标测定结果的异常值,并根据模型主要参数:内外部验证决定系数(r2)、校正标准偏差(rmsec)、预测标准偏差(rmsep)等,进行模型质量评价。具体计算公式如下:
式中,yi,actual为第i样本参考方法的测定值;
式中,yi,actual为第i样本参考方法的测定值;yi,predicted为用所建立模型对校正集中第i样本的预测值;n为校正集的样本数。rmsec值越小越好。
式中,yi,actual为第i样本参考方法的测定值;yi,predicted为验证集预测过程中第i样本的光谱方法预测值;m为验证集的样本数。rmsep值越小越好,
rmsec应小于rmsep值。
结果nir光谱模型如图7~图9显示,颈腰康胶囊士的宁nir模型中,内部验证直线方法为y=0.9731x+0.0184,外部验证直线方程为y=0.9614x+0.0266,其中x为化验值,y为预测值,内部验证决定系数r2=0.9865,外部验证决定系数r2=0.9778,rmsec=0.0116,rmsep=0.0149;颈腰康胶囊的羟基红花黄色素anir模型中,内部验证直线方法为y=0.9547x+0.0330,外部验证直线方程为y=0.9317x+0.0500,内部验证决定系数r2=0.9771,外部验证决定系数r2=0.9638,rmsec=0.0192,rmsep=0.0241;颈腰康胶囊水分nir模型中,内部验证直线方法为y=0.9750x+0.1464,外部验证直线方程为y=0.9469x+0.3104,内部验证决定系数r2=0.9874,外部验证决定系数r2=0.9750,rmsec=0.1290,rmsep=0.1813。
上述参数表明,参与建模的颈腰康胶囊各指标含量与其nir光谱相关性较好,所建立的nir检测模型性能良好。
实施例5模型验证
利用已建立的各指标校正模型,对未参与建模的样品进行预测分析,验证样品与建模样品光谱采集参数设置的一致性,获得各指标的预测结果,计算预测值与实测值偏差,验证模型准确性。偏差值计算公式如下:
表3颈腰康胶囊nir模型验证结果
结果可知,5批次验证样品中士的宁、羟基红花黄色素a及水分含量预测值与实测值平均偏差值分别为2.75%、2.88%、2.85%,预测偏差值均小于5%。由此可见,各指标nir检测模型预侧值与常规检测方法测定值之间相关性良好,测定准确性较高,可以采用nir光谱模型快速检测分析颈腰康胶囊中的士的宁、羟基红花黄色素a及水分含量。
由此可见,各指标nir检测模型预侧值与常规检测方法测定值之间相关性良好,测定准确性较高,可以采用nir光谱模型快速检测颈腰康胶囊中的士的宁、羟基红花黄色素a及水分含量。
实施例6与原有检测技术对比评价
对已建立的士的宁、羟基红花黄色素a及水分定量模型,分别采用5批未参与建模的样品进行外部验证,结果证明nir技术预测结果与传统高效液相色谱法测定结果基本一致,无明显偏差。nir快速检测准确度较高,可替代液相分析。在应用及相关耗材方面也进行了对比,具体见表4.
表4检测技术对比
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!