基于碳量子点的荧光免疫分析方法与流程
本发明涉及抗原检测技术领域,具体地说,涉及基于碳量子点的荧光免疫分析方法。
背景技术:
免疫分析是基于抗原与抗体之间特异性识别和结合的一类快速分析技术。免疫分析可适用于大分子化合物(如蛋白质、细菌等)和小分子化合物(如农药、兽药、真菌毒素等)的测定。由于免疫分析具有高特异性、检测速度快、低成本和适于大量样品现场筛选等优点,使它在食品安全检测、环境监测等分析领域中得到广泛应用。
酶联免疫分析方法是较为常用的一种免疫分析方法,具有仪器依赖度低、操作简便、低成本、特异性强、灵敏度较高、可实现大批量检测、易商品化等优点,已经成为市场上一种应用广泛,成熟的检测与分析技术,并有大量的相关的试剂盒产品投放市场。食品中小分子化学污染物因其分子量较小(≤6000da),往往只有单一抗原决定簇,因而通常采用竞争酶联免疫分析法来对其进行定量检测。传统的竞争酶联免疫分析法通常采用辣根过氧化物酶或碱性磷酸酶催化底物显色,并以吸光度值的形式作为信号输出。然而,酶催化底物产生的吸光度信号通常具有较低的信噪比,导致酶联免疫分析的灵敏度较低。
为了达到更好的检测灵敏度,建立具有高信噪比的荧光免疫分析方法是一种有效的策略。传统荧光染料试剂有着荧光稳定性差,不耐光漂白,斯托克斯位移小等缺陷,因而限制了其在荧光免疫分析方法中的应用。传统的荧光纳米材料如cdse等半导体量子点、荧光微球、上转换纳米颗粒、稀土纳米荧光配合物等通常具有量子产率高,荧光稳定性好,激发谱较宽、斯托克斯位移大等优良的光学特性,常被用于荧光免疫分析。然而半导体量子点等传统的荧光纳米材料通常合成条件苛刻,合成步骤复杂,含有重金属物质,存在一定的生物毒害性。而且半导体量子点等必须经过复杂的“油-水”转相和表面修饰才能应用于荧光免疫分析中。以上这些因素限制了传统的荧光纳米材料的应用,因此在荧光免疫分析中,光学性能优异,合成简便,成本低,稳定好,无毒害作用的新型荧光探针的开发具有重要的意义。
碳量子点是一种新型的碳基荧光纳米材料,具有优异的光学性能,良好的水溶性、不含有毒元素、环境友好、合成原料来源广、成本低、合成简便、生物相容性好等诸多优点。这些特性使碳量子点在生物分析和生物传感等方面具有广阔的应用前景。目前碳点在免疫分析中的应用较少,其在免疫分析中的应用潜力尚有待开发。
技术实现要素:
本发明的目的是提供一种基于碳量子点的荧光免疫分析方法。
为了实现本发明目的,第一方面,本发明提供一种荧光碳量子点,所述碳量子点是以柠檬酸为碳源,硫脲或尿素为氮源制备得到的。具体地,所述荧光碳量子点按如下方案i或ii制备得到:
方案i:将3-5g柠檬酸和5-7g硫脲加入到15-20ml水中,搅拌使化合物溶解,将混合液置于微波反应器中,800-1000w功率下加热7-10min,直至溶液从无色变为深棕色固体,然后冷却至室温,加入25ml水溶解,所得碳量子点溶液经过滤、透析及色谱柱纯化,即得;
方案ii:将3-5g柠檬酸和5-7g尿素加入到15ml水中,搅拌使化合物溶解,将混合液置于微波反应器中,800-1000w功率下加热7-10min,直至溶液从无色变为深棕色固体,然后冷却至室温,加入25ml水溶解,所得碳量子点溶液经过滤、透析及色谱柱纯化,即得。
优选地,所述荧光碳量子点按如下方案i′或ii′制备得到:
方案i′:将5g柠檬酸和7g硫脲加入到15ml水中,搅拌使化合物溶解,将混合液置于微波反应器中,800w功率下加热7分钟,直至溶液从无色变为深棕色固体,然后冷却至室温,加入25ml水溶解,所得碳量子点溶液经过滤、透析及色谱柱纯化,即得;
方案ii′:将3g柠檬酸和5g尿素加入到15ml水中,搅拌使化合物溶解,将混合液置于微波反应器中,800w功率下加热7分钟,直至溶液从无色变为深棕色固体,然后冷却至室温,加入25ml水溶解,所得碳量子点溶液经过滤、透析及色谱柱纯化,即得。
前述方法中,所述过滤优选采用0.22μm膜过滤。
所述透析优选使用型号为3500da的透析膜在超纯水中透析至少24h;和/或
所述色谱柱优选为sephadexg25柱。
按照方案i制备的荧光碳量子点(cds-1)的激发波长为370nm-380nm,发射波长为520nm-530nm。370nm激发,520nm发射时,cds-1绝对量子产率为32.6%。
按照方案ii制备的荧光碳量子点(cds-2)的激发波长为400nm-410nm,发射波长为515nm-525nm。400nm激发,515nm发射时,cds-2绝对量子产率为27.1%。
第二方面,本发明提供所述荧光碳量子点的以下任一应用:
①制备生物探针、生物传感器以及催化领域中的应用;
②在免疫分析中的应用。
第三方面,本发明提供由所述荧光碳量子点制备的生物探针、生物传感器以及检测试剂、试剂盒。
第四方面,本发明提供基于碳量子点的荧光免疫分析方法,包括以下步骤:
s1、用人工抗原包被酶标板的各反应孔;
s2、向反应孔中加入特异性抗体和待测样品溶液,孵育一段时间后,向反应孔中加入孔辣根过氧化物酶或碱性磷酸酶标记的二抗,继续孵育;
s3、向反应孔中加入辣根过氧化物酶底物3,3’,5,5’-四甲基联苯胺溶液或碱性磷酸酶底物4-硝基苯磷酸二钠溶液,进行显色反应;
s4、显色反应结束后,向反应孔中加入方案i或ii制备的所述荧光碳量子点,检测荧光强度;
s5、配制不同浓度的小分子半抗原标准品溶液,按照s2-s4进行检测,建立反映荧光强度与小分子半抗原浓度之间关系的标准曲线;
s6、根据待测样品溶液的检测结果,对照标准曲线,获得待测样品溶液中小分子半抗原的浓度。
本发明的检测原理如下:辣根过氧化物酶在h2o2的存在下,可催化氧化3,3’,5,5’-四甲基联苯胺(tmb)生成四甲基联苯胺氧化聚合物(tmbox)。tmbox是一种强吸光物质,它的吸收光谱与cds-1的激发光谱有很好的重叠,从而产生明显的荧光内滤效应,可以有效地猝灭cds-1的荧光。类似的原理,碱性磷酸酶催化对硝基苯磷酸二钠(pnpp)水解产生的对硝基酚(pnp)也是一种强吸光物质,它的吸收光谱与cds-2的激发光谱有很好的重叠,产生明显的荧光内滤效应,从而可以有效地猝灭cds-2的荧光。通过监测cds-1/cds-2的荧光猝灭率,可以成功地将酶联免疫分析中辣根过氧化物酶/碱性磷酸酶催化底物反应形成的吸光度信号有效地转化为荧光信号,从而增大了检测窗口,提高了检测信噪比,并显著地提高了酶联免疫分析的检测灵敏度。
在本发明的一个具体实施方式中,所述荧光免疫分析方法包括以下步骤(以金刚烷胺荧光免疫分析为例):
1)用包被液将(金刚烷胺)人工抗原稀释到浓度为3.3×10–5mg/ml,按100μl/孔加入96孔酶标板中,4℃孵育12h后,以洗涤液洗板;
2)按150μl/孔加入封闭液进行封闭,37℃孵育1h,以洗涤液洗板;
3)按50μl/孔加入特异性(金刚烷胺)抗体(浓度为2.5×10–5mg/ml)和按50μl/孔加入待测样品溶液,37℃孵育0.5h,抗原-抗体发生特异性结合反应,再以洗涤液洗板;
4)按100μl/孔加入辣根过氧化物酶或碱性磷酸酶标记的二抗(浓度为0.1mg/ml),37℃孵育0.5h,形成酶标记免疫复合物,以洗涤液洗板;
5)按100μl/孔将辣根过氧化物酶底物3,3’,5,5’-四甲基联苯胺溶液(浓度为2.5mm)或碱性磷酸酶底物4-硝基苯磷酸二钠溶液(浓度为5mm)分别加入相应的酶标板孔中,37℃孵育20min;
6)按20μl/孔加入20μg/ml的碳量子点溶液,并用多功能酶标仪检测荧光值;
具体地,将cds-2溶液加入步骤5)中辣根过氧化物酶催化显色反应结束后的酶标板孔中(或将cds-2溶液加入步骤5)中碱性磷酸酶催化显色反应结束后的酶标板孔中),并用多功能酶标仪检测荧光值;设定激发波长370nm,扫描400-600nm处cds-1的荧光光谱,读取520nm处荧光值(或激发波长400nm,扫描420-600nm处cds-2的荧光光谱,读取515nm处荧光值);
7)配制不同浓度梯度的小分子半抗原标准品溶液,按照3)-6)进行检测,处理所得数据,绘制标准曲线;
8)根据待测样品溶液的检测结果,对照标准曲线,获得待测样品溶液中小分子半抗原的浓度;
优选地,所述包被液为:10mm、ph9.6的碳酸盐缓冲液。
所述洗涤液为:含0.05%tween-20的50mm、ph7.4的磷酸盐缓冲液。
所述封闭液为:含2%bsa的磷酸盐缓冲液。
本发明所述小分子半抗原来自真菌毒素、农药、兽药或环境激素。例如,所述小分子半抗原包括但不限于金刚烷胺、黄曲霉毒素b1或利巴韦林。
所述人工抗原为所述小分子半抗原与牛血清白蛋白的偶联物。例如,金刚烷胺-牛血清白蛋白偶联物、黄曲霉毒素b1-牛血清白蛋白偶联物或利巴韦林-牛血清白蛋白偶联物。
所述特异性抗体为抗所述小分子半抗原的单克隆抗体。例如,抗金刚烷胺鼠源单克隆抗体、抗黄曲霉毒素b1单克隆抗体或利巴韦林单克隆抗体。
利用上述方法可实现样品中小分子半抗原的定量检测。当所述小分子半抗原为金刚烷胺、黄曲霉毒素b1或利巴韦林时,相应的线性检测范围及最低检测限分别如下:
金刚烷胺的线性检测范围:0.035-1.1ng/ml,最低检测限:0.02ng/ml。
黄曲霉毒素b1的线性检测范围:0.016-0.95ng/ml,最低检测限:0.012ng/ml。
利巴韦林的线性检测范围:0.18-0.74ng/ml,最低检测限:0.015ng/ml。
本发明中,所述抗金刚烷胺鼠源单克隆抗体、抗黄曲霉毒素b1单克隆抗体及利巴韦林单克隆抗体均由中国农业大学国家兽药安全评价中心提供,制备方法可参见:
wang,z.,wen,k.,zhang,x.,li,x.,wang,z.,shen,j.,ding,s.(2018).newhaptensynthesis,antibodyproduction,andindirectcompetitiveenzyme-linkedimmnunosorbentassayforamantadineinchickenmuscle.foodanalyticalmethods,11(1),302-308.
zhang,x.,song,m.,yu,x.,wang,z.,ke,y.,jiang,h.,wen,k.(2017).developmentofanewbroad-specificmonoclonalantibodywithuniformaffinityforaflatoxinsandmagneticbeads-basedenzymaticimmunoassay.foodcontrol,79,309-316.
wang,z.,yu,x.,ma,l.,liu,h.,ding,s.,wang,z.,wen,k.(2018).preparationofhighaffinityantibodyforribavirinwithnewhaptensandresidueanalysisinchickenmuscle,eggsandduckmuscle.foodadditives&contaminants:parta,35,1247-1256.
借由上述技术方案,本发明至少具有下列优点及有益效果:
(一)本发明提供了一种基于碳量子点的高灵敏度的荧光免疫分析新方法,具有操作简单、成本低、灵敏度高和稳定性好等优点。
(二)碳量子点无需额外的表面功能化修饰,可直接作为荧光探针应用于酶联免疫分析中,简化了操作。
(三)利用碳量子点与酶催化的显色底物间产生高效的荧光内滤效应,可以成功地将酶联免疫分析中辣根过氧化物酶/碱性磷酸酶催化底物反应形成的吸光度信号有效地转化为荧光信号,具有良好扩展性,有望扩宽至其它目标检测体系。
(四)碳量子点荧光量子产率高、发光稳定、合成简便、无毒性,用作检测探针在可控性上稳定、操作简便、成本低廉、对环境友好。
(五)本发明适用于小分子半抗原的定量检测,如真菌毒素、农药、兽药、环境激素等,样品处理按照国家标准检测处理方法即可。通过改变使用识别抗体和相应的人工抗原,利用碳量子点作为荧光探针,本方法可扩展到蛋白质、核酸等目标物的非竞争酶联免疫分析检测中。
(六)本发明至少成功解决了以下技术问题:①现有酶联免疫分析方法中因显色底物信噪比较低而导致检测灵敏度较低的技术问题;②常规的通过碳量子点替代酶作为信号探针,应用于小分子半抗原检测的竞争免疫分析方法中,碳量子点与抗体的偶联效率较低,偶联效果难以得到保证;③当采用碳量子点作为荧光信号探针,应用于小分子半抗原检测的竞争免疫分析方法中时,往往需要对碳量子点进行复杂的表面功能化修饰,修饰效果难以得到保证。
附图说明
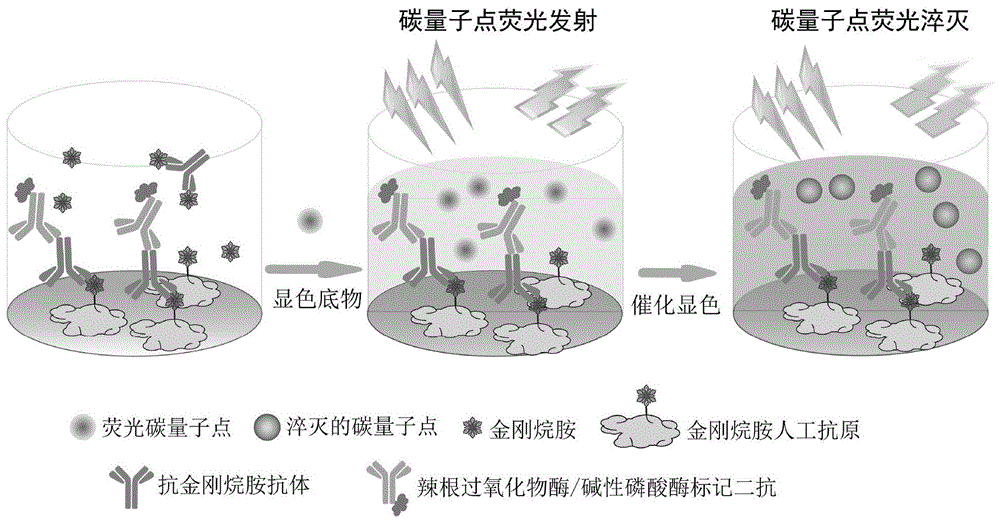
图1为本发明实施例2中检测金刚烷胺的荧光酶联免疫分析方法原理示意图。
图2为本发明实施例1中合成的碳量子点的荧光光谱图。
图3为本发明实施例1中合成的碳量子点的透射电镜图。
图4分别为本发明实施例2-4中检测金刚烷胺、黄曲霉毒素b1和利巴韦林浓度的检测标准曲线图。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段,所用的试验试剂耗材,如无特殊说明,均为常规生化试剂。
以下实施例中涉及的定量试验,均设置三次重复实验,结果取平均值。
以下实施例中,磷酸盐缓冲液(pbs,0.05m,ph7.4)的配制如下:将nacl40g,na2hpo413.5g,kh2po41.0g,kcl1.0g溶于1l超纯水中。
洗涤液:0.05m,pbs(ph7.4)+0.05%(w/w)tween-20。
封闭液:0.05m,pbs(ph7.4)+1%(w/w)bsa溶液。
包被液:碳酸盐缓冲液(cb,0.01m、ph9.6),将na2co31.59g和nahco32.94g加超纯水定容至1l。
以下实施例中所涉及的所有小分子半抗原标准品与常规生化试剂等均购买自sigma公司。
实施例1荧光碳量子点的合成
将5g柠檬酸和7g硫脲加入到15ml水中。不断搅拌使化合物溶解,将混合溶液置于微波反应器中,800w功率下加热7min,直至溶液从无色变为深棕色固体,表明反应物碳化,有碳量子点生成。冷却至室温后,加入25ml水以溶解产物。将所制备的碳量子点通过0.22μm膜过滤以除去大颗粒,并使用透析膜(3500da)在超纯水透析至少24h以除去未反应的小分子。使用sephadexg25柱色谱法对制备的碳量子点溶液进行分离和纯化,收集得到的最大激发波长为370nm,最大发射波长为520nm的碳量子点溶液,命名为cds-1。
将3g柠檬酸和5g尿素加入到15ml超纯水中以形成透明溶液。不断搅拌使化合物溶解,将混合溶液置于微波反应器中,800w功率下加热7min,直至溶液从无色变为深棕色固体,表明反应物碳化,有碳量子点生成。冷却至室温后,加入25ml水以溶解产物。将所制备的碳量子点通过0.22μm膜过滤以除去大颗粒,并使用透析膜(3500da)在超纯水透析至少24h以除去未反应的小分子。使用sephadexg25柱色谱法对制备的碳量子点溶液进行分离和纯化,收集得到的最大激发波长为400nm,最大发射波长为515nm的碳量子点溶液,命名为cds-2。
合成的碳量子点的荧光光谱图见图2,透射电镜图见图3。
实施例2金刚烷胺荧光免疫分析检测
1)用包被液将金刚烷胺人工抗原稀释到浓度为3.3×10–5mg/ml,按100μl/孔加入96孔酶标板中,4℃孵育12h后,以洗涤液洗板;
2)按150μl/孔加入封闭液进行封闭,37℃孵育1h,以洗涤液洗板;
3)按50μl/孔加入特异性抗金刚烷胺抗体(浓度为2.5×10–5mg/ml)和按50μl/孔加入待测样品溶液,37℃孵育0.5h,抗原-抗体发生特异性结合反应,再以洗涤液洗板;
4)按100μl/孔加入辣根过氧化物酶或碱性磷酸酶标记的二抗(浓度为0.1mg/ml),37℃孵育0.5h,形成酶标记免疫复合物,以洗涤液洗板;
5)按100μl/孔将辣根过氧化物酶底物3,3’,5,5’-四甲基联苯胺溶液(浓度为2.5mm)或碱性磷酸酶底物4-硝基苯磷酸二钠溶液(浓度为5mm)分别加入相应的酶标板孔中,37℃孵育20min;
6)信号的转换与放大:20μl/孔20μg/ml的碳量子点cds-1溶液加入步骤5)中辣根过氧化物酶催化显色反应结束后的酶标板孔中(或cds-2溶液加入步骤5)中碱性磷酸酶催化显色反应结束后的酶标板孔中);并用perkinelmerenvision多功能酶标仪检测荧光值,设定激发波长370nm,扫描400-600nm处cds-1的荧光光谱,读取520nm处荧光值(或激发波长400nm,扫描420-600nm处cds-2的荧光光谱,读取515nm处荧光值)。
7)荧光免疫分析标准曲线建立:已知浓度梯度的金刚烷胺标准品按照步骤3)-6)方法进行检测,处理步骤6)所得数据,绘制标准曲线;获得的标准曲线如图4(a和b)所示,随着金刚烷胺浓度增加,碳量子点荧光猝灭率下降,强度逐渐恢复。通过标准曲线计算得该方法的金刚烷胺线性检测范围为0.035-1.1ng/ml,检测线即ic10为0.02ng/ml。
本发明检测金刚烷胺的荧光酶联免疫分析方法原理示意图见图1。检测金刚烷胺浓度的检测标准曲线见图4-a和图4-b。其中,图4-a为基于辣根过氧化物酶的金刚烷胺荧光酶联免疫分析标准曲线,图4-b为基于碱性磷酸酶的金刚烷胺荧光酶联免疫分析标准曲线。
传统的酶联免疫分析按照步骤1)-5)的方法进行检测,酶促反应完成后,每孔加入50μl2mol/l的浓硫酸溶液(针对辣根过氧化物酶催化的显色底物),以450nm波长处吸光度测定各孔od值;或者,酶促反应完成后,每孔加入2mol/lnaoh溶液(针对碱性磷酸酶催化的显色底物),以405nm波长处吸光度测定各孔od值。已知浓度梯度的金刚烷胺标准品按照步骤1)-5)方法进行检测,处理步骤5)所得数据,绘制标准曲线;通过标准曲线计算得该方法的线性检测范围为为0.23-9.1ng/ml,检测线即ic10为0.2ng/ml。
实施例3黄曲霉毒素b1荧光免疫分析检测
1)用包被液将黄曲霉毒素b1人工抗原稀释到浓度为2.3×10–5mg/ml,按100μl/孔加入96孔酶标板中,4℃孵育12h后,以洗涤液洗板;
2)按150μl/孔加入封闭液进行封闭,37℃孵育1h,以洗涤液洗板;
3)按50μl/孔加入特异性抗黄曲霉毒素b1抗体(浓度为2.1×10–5mg/ml)和按50μl/孔加入待测样品溶液,37℃孵育0.5h,抗原-抗体发生特异性结合反应,再以洗涤液洗板;
4)按100μl/孔加入辣根过氧化物酶或碱性磷酸酶标记的二抗(浓度为0.1mg/ml),37℃孵育0.5h,形成酶标记免疫复合物,以洗涤液洗板;
5)按100μl/孔将辣根过氧化物酶底物3,3’,5,5’-四甲基联苯胺溶液(浓度为2.5mm)或碱性磷酸酶底物4-硝基苯磷酸二钠溶液(浓度为5mm)分别加入相应的酶标板孔中,37℃孵育20min;
6)信号的转换与放大:20μl/孔20μg/ml的碳量子点cds-1溶液加入步骤5)中辣根过氧化物酶催化显色反应结束后的酶标板孔中(或cds-2溶液加入步骤5)中碱性磷酸酶催化显色反应结束后的酶标板孔中);并用perkinelmerenvision多功能酶标仪检测荧光值,设定激发波长370nm,扫描400-600nm处cds-1的荧光光谱,读取520nm处荧光值(或激发波长400nm,扫描420-600nm处cds-2的荧光光谱,读取515nm处荧光值)。
7)荧光免疫分析标准曲线建立:已知浓度梯度的黄曲霉毒素b1标准品按照步骤3)-6)方法进行检测,处理步骤6)所得数据,绘制标准曲线;获得的标准曲线如图4(c和d)所示,随着黄曲霉毒素b1浓度增加,碳量子点荧光猝灭率下降,强度逐渐恢复。通过标准曲线计算得该方法的线性检测范围为0.016-0.95ng/ml,检测线即ic10为0.012ng/ml。其中,图4-c为基于辣根过氧化物酶的黄曲霉毒素b1荧光酶联免疫分析标准曲线,图4-d为基于碱性磷酸酶的黄曲霉毒素b1荧光酶联免疫分析标准曲线。
传统的酶联免疫分析按照步骤1)-5)的方法进行检测,酶促反应完成后,每孔加入50μl2mol/l的浓硫酸溶液(针对辣根过氧化物酶催化的显色底物),以450nm波长处吸光度测定各孔od值;或者,酶促反应完成后,每孔加入2mol/lnaoh溶液(针对碱性磷酸酶催化的显色底物),以405nm波长处吸光度测定各孔od值。已知浓度梯度的金刚烷胺标准品按照步骤1)-5)方法进行检测,处理步骤5)所得数据,绘制标准曲线;通过标准曲线计算得该方法的线性检测范围为0.14-3.75ng/ml,检测线即ic10为0.1ng/ml。
实施例4利巴韦林荧光免疫分析检测
1)用包被液将利巴韦林人工抗原稀释到浓度为3.0×10–5mg/ml,按100μl/孔加入96孔酶标板中,4℃孵育12h后,以洗涤液洗板;
2)按150μl/孔加入封闭液进行封闭,37℃孵育1h,以洗涤液洗板;
3)按50μl/孔加入特异性抗利巴韦林抗体(浓度为1.5×10–5mg/ml)和按50μl/孔加入待测样品溶液,37℃孵育0.5h,抗原-抗体发生特异性结合反应,再以洗涤液洗板;
4)按100μl/孔加入辣根过氧化物酶或碱性磷酸酶标记的二抗(浓度为0.1mg/ml),37℃孵育0.5h,形成酶标记免疫复合物,以洗涤液洗板;
5)按100μl/孔将辣根过氧化物酶底物3,3’,5,5’-四甲基联苯胺溶液(浓度为2.5mm)或碱性磷酸酶底物4-硝基苯磷酸二钠溶液(浓度为5mm)分别加入相应的酶标板孔中,37℃孵育20min;
6)信号的转换与放大:20μl/孔20μg/ml的碳量子点cds-1溶液加入步骤(5)中辣根过氧化物酶催化显色反应结束后的酶标板孔中(或cds-2溶液加入步骤(5)中碱性磷酸酶催化显色反应结束后的酶标板孔中);并用perkinelmerenvision多功能酶标仪检测荧光值,设定激发波长370nm,扫描400-600nm处cds-1的荧光光谱,读取520nm处荧光值(或激发波长400nm,扫描420-600nm处cds-2的荧光光谱,读取515nm处荧光值)。
7)荧光免疫分析标准曲线建立:已知浓度梯度的利巴韦林标准品按照步骤3)-6)方法进行检测,处理步骤6)所得数据,绘制标准曲线;获得的标准曲线如图4(e和f)所示,随着利巴韦林浓度增加,碳量子点荧光猝灭率下降,强度逐渐恢复。通过标准曲线计算得该方法的线性检测范围为0.18-0.74ng/ml,检测线即ic10为0.015ng/ml。其中,图4-e为基于辣根过氧化物酶的利巴韦林荧光酶联免疫分析标准曲线,图4-f为基于碱性磷酸酶的利巴韦林荧光酶联免疫分析标准曲线。
传统的酶联免疫分析按照步骤1)-5)的方法进行检测,酶促反应完成后,每孔加入50μl2mol/l的浓硫酸溶液(针对辣根过氧化物酶催化的显色底物),以450nm波长处吸光度测定各孔od值;或者,酶促反应完成后,每孔加入2mol/lnaoh溶液(针对碱性磷酸酶催化的显色底物),以405nm波长处吸光度测定各孔od值。已知浓度梯度的利巴韦林标准品按照步骤1)-5)方法进行检测,处理步骤5)所得数据,绘制标准曲线;通过标准曲线计算得该方法的线性检测范围为0.23ng/ml-8.25ng/ml,检测线即ic10为0.15ng/ml。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之做一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!