液相色谱-高分辨质谱法用于人体尿液中吗啡的测定方法与流程
本发明涉及毒品检测领域,即液相色谱-高分辨质谱法用于人体尿液中吗啡的测定方法。
背景技术:
在现有技术中,关于人体尿液中吗啡的测定,国内目前常用的检测方法有气相色谱法、气相色谱-质谱法等,存在的缺点有:1.气相色谱法仅能对吗啡进行定量分析,根据目标峰的保留时间粗略判断,存在假阳性的风险,而且气相色谱法的灵敏度较低,仅能对mg/l级别进行定量分析,根据ga/t1318-2016提供的对吸毒人员尿液中吗啡的最低检出限为300mg/l。2.气相色谱-质谱法能够对吗啡定性和定量分析,但同样存在灵敏度低的缺点,根据ga/t1318-2016提供的对吸毒人员尿液中吗啡的最低检出限为50mg/l。
总之,原有的人体尿液中吗啡的测定采用的检测方法,前处理过程繁琐,检测灵敏度低,对于含量低的样品常发生假阳性或者未检出的情况。
技术实现要素:
本发明的目的是提供一种液相色谱-高分辨质谱法用于人体尿液中吗啡的测定方法,能够对人体尿液中痕量吗啡的定性和定量测定,检测更准确、方便、灵敏度高、精度高。
本发明的技术解决方案是:液相色谱-高分辨质谱法用于人体尿液中吗啡的测定方法,其特征在于步骤如下:
人体尿液中的吗啡经hlb固相萃取柱净化,用液相色谱-高分辨质谱仪测定,外标法定量。
a、试样溶液制备:量取人体尿液与水各10.0ml,混匀。
b、吗啡的提取、净化、洗脱和浓缩:将2.00ml样液加入到hlb固相萃取柱中,流出液弃去;依次加入5ml碳酸氢钠-硼酸缓冲溶液、5ml水淋洗,流出液弃去;待空气通过hlb固相萃取柱3min;加入10ml乙腈-甲醇溶液(9:1,v/v)溶液洗脱,流出速度为1ml/min,用鸡心瓶收集全部流出液;鸡心瓶在旋转蒸发仪45℃水浴中旋至近干,在氮吹仪上使用氮气吹干;用1ml定容液溶解残渣,过膜得到试样溶液,供液相色谱-高分辨质谱仪测定。
c、液相色谱参考条件:色谱柱:accucoreaq柱,150mm×2.1mm,2.6μm或性能相当者;流动相a:甲酸-甲酸铵溶液,流动相b:乙腈,流速0.3ml/min,进样量5μl,梯度洗脱为:
d、高分辨质谱检测:
电离模式:正离子模式。
喷雾电压:3.0kv。
加热器温度:50℃。
鞘气:40arb,氮气。
辅助气:5arb,氮气。
扫描模式:一级全扫描/数据依赖二级子离子扫描。
采集范围:m/z100~m/z500。
一级全扫描:分辨率:70000fwhm。
数据依赖二级子离子扫描:分辨率:17500fwhm。
所述的甲酸-甲酸铵溶液是量取1.0ml甲酸,用水溶解,作为溶液a;称取0.63g甲酸铵,用水溶解,作为溶液b;将溶液a和溶液b混匀,定容至1.0l,配制成含有0.1%甲酸和10mmol/l甲酸铵的水溶液。
本发明的优点是:1、本方法不受分析物挥发性、极性、热稳定性的限制,无需衍生,而且多反应监测模式能锁定目标物的母离子、子离子对,有效降低噪音干扰,因此应用范围更广,检测更准确、方便,灵敏度更高。2、原有的人体尿液中吗啡的测定采用的检测方法,前处理过程繁琐,检测灵敏度低,对于含量低的样品常发生假阳性或者未检出的情况。本方法前处理简单,经济成本低,消耗人力物力少,操作过程简单,采用的液相色谱-高分辨质谱法,是目前国际上尖端的检测方法,检测精度、灵敏度、准确度都远远高于原有的气相色谱法和气相色谱-质谱法,测定低限1μg/l,高于气相色谱-质谱法检出限50mg/l五万倍,能够对人体尿液中超痕量吗啡的定性和定量测定。3、根据下面操作步骤5.1和5.2可知,该方法的前处理简便,使用试剂较少,无需衍生,大量地节省人力和经济成本。根据下面操作步骤5.3可知,定量和定性方法操作性强,灵敏度、准确度和仪器精度更高,根据下面操作步骤8测定低限1.0μg/l可知,是目前所有检测方法中最低的,而且减少假阳性检出的风险。
下面将结合实施例对本发明的实施方式作进一步详细描述。
附图说明
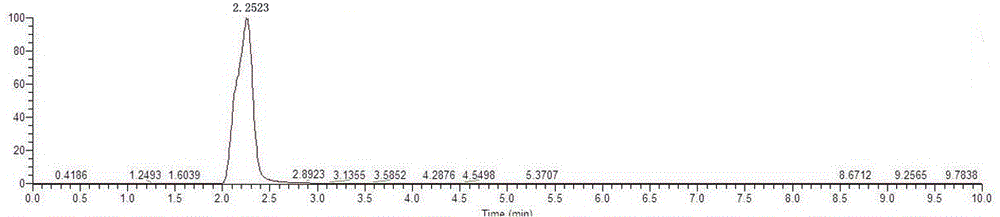
图1是吗啡标准溶液色谱图。
图2是吗啡标准溶液的一级全扫描(fullms)质谱图。
图3是吗啡标准溶液的数据依赖二级子离子扫描(dd-ms2)质谱图。
图4是吗啡的标准曲线。
具体实施方式
液相色谱-高分辨质谱法用于人体尿液中吗啡的测定方法
1.原理:人体尿液中的吗啡经hlb固相萃取柱净化,用液相色谱-高分辨质谱仪测定,外标法定量。
2.试剂和材料
2.1甲酸铵(hcoonh4):cas号540-69-2。
2.2碳酸氢钠(na2co3):cas号144-55-8。
2.3硼酸(h3bo3):cas号10043-35-3。
2.4氢氧化钠(naoh):cas号1310-73-2。
2.5乙腈(c2h3n):色谱纯,cas号75-05-8。
2.6甲醇(ch3oh):色谱纯,cas号67-56-1。
2.7甲酸(ch2o2):色谱纯,cas号64-18-6。
2.8乙腈-甲醇溶液:乙腈(2.5)和甲醇(2.6)(9+1,v+v),混匀。
2.9碳酸氢钠溶液:称取8.4g碳酸氢钠(2.2),以水溶解,定容至1.0l,配制成0.1mol/l碳酸氢钠溶液,用塑料容器盛装。
2.10硼酸溶液:称取6.2g硼酸(2.3),以水溶解,定容至1.0l,配制成0.1mol/l硼酸溶液。
2.11氢氧化钠溶液:称取4.0g氢氧化钠(2.4),以水溶解,定容至1.0l,配制成0.1mol/l氢氧化钠溶液,用塑料容器盛装。
2.12碳酸氢钠-硼酸缓冲溶液:将碳酸氢钠溶液(2.9)和硼酸溶液(2.10)等体积混和,由氢氧化钠溶液(2.11)调节ph值为9.5,用ph计(2.2)测定ph值,用塑料容器盛装。
2.13甲酸-甲酸铵溶液:量取1.0ml甲酸(2.7),用少量水溶解,作为溶液a;称取0.63g甲酸铵(2.1),用少量水溶解,作为溶液b。将溶液a和溶液b混匀,定容至1.0l,配制成含有0.1%甲酸和10mmol/l甲酸铵的水溶液。
2.14定容液:乙腈(2.5)和甲酸-甲酸铵溶液(2.13)(5+95,v+v),混匀。
2.15吗啡标准物质:吗啡标准溶液1.0mg/ml,cas号57-27-2。
2.16吗啡标准储备液:准确吸取1.0ml吗啡标准溶液(2.15)置于10ml棕色容量瓶中,以甲醇(2.6)溶解,并定容至刻度,配制成吗啡浓度为100.0μg/l的标准储备溶液,0~4℃保存,备用,有效期为6个月。
2.17吗啡标准工作液:准确移取适量的吗啡标准储备液(2.16),用定容液(2.14)稀释成合适浓度的吗啡标准工作液,现用现配。
2.18亲水-亲酯固相萃取柱(hlb固相萃取柱):6ml,500mg或相当者,使用前依次用1ml甲醇(2.6)、1ml水、1ml碳酸氢钠-硼酸缓冲溶液(2.12)活化。
2.19有机相微孔滤膜:0.22μm。
3.仪器与设备
3.1液相色谱-高分辨质谱仪:配有电喷雾离子源(esi)。
3.2ph计。
3.3旋转蒸发仪。
3.4氮吹仪。
3.5电子天平:感量0.01g和0.001g。
3.6涡旋混合器。
4.样品
4.1样品储存:采集人体尿液立即测试,若不能及时测试,装于玻璃容器中,在-18℃以下冷冻保存,需测试时待样品缓至室温再进行操作。
4.2样液:量取人体尿液与水各10.0ml,混匀。
5.操作步骤
5.1吗啡的提取、净化、洗脱和浓缩:将2.00ml样液(4.2)加入到hlb固相萃取柱(2.18)中,流出液弃去。依次加入5ml碳酸氢钠-硼酸缓冲溶液(2.12)、5ml水淋洗,流出液弃去。待空气通过hlb固相萃取柱3min。加入10ml乙腈-甲醇溶液(2.8)洗脱,流出速度为1ml/min,用鸡心瓶收集全部流出液。鸡心瓶在旋转蒸发仪(3.3)45℃水浴中旋至近干,在氮吹仪(3.4)上使用氮气吹干。用1ml定容液(2.14)溶解残渣,过膜得到试样溶液,供液相色谱-高分辨质谱仪(3.1)测定。
此步骤为人体尿液中吗啡的主要前处理方法,相比较于以往的方法,无需衍生,仅用一个固相萃取柱提取、洗脱即可,操作方法简单,所需化学试剂少,降低了经济和人力成本,减少了化学试剂对操作人的潜在伤害和对环境的污染。
5.2空白试验:不添加试样,按上述步骤5.1操作。
5.3测定
5.3.1液相色谱参考条件
5.3.1.1色谱柱:accucoreaq柱,150mm×2.1mm,2.6μm或性能相当者;
5.3.1.2流动相:a:甲酸-甲酸铵溶液(2.13),b:乙腈(2.5),梯度洗脱条件参见表1;
5.3.1.3流速:0.3ml/min;
5.3.1.4进样量:5μl。
5.3.2高分辨质谱参考条件
5.3.2.1电离模式:正离子模式;
5.3.2.2喷雾电压:3.0kv;
5.3.2.3加热器温度:50℃;
5.3.2.4鞘气:40arb,氮气;
5.3.2.5辅助气:5arb,氮气;
5.3.2.6扫描模式:一级全扫描/数据依赖二级子离子扫描(fullms/dd-ms2);
5.3.2.7采集范围:m/z100~m/z500;
5.3.2.8一级全扫描(fullms):分辨率:70000fwhm;
5.3.2.9数据依赖二级子离子扫描(dd-ms2):分辨率:17500fwhm。
5.3.3定性分析
按照5.3.1和5.3.2的仪器条件测定试样溶液和标准工作液,如果试样溶液与标准工作液中目标物的保留时间一致,保留时间偏差在±2.5%以内,母离子的测量质核比与表2中母离子的精确质核比数值相差值与精确质量数的比值小于5×10-6,并同时测得表2所示的特征离子,可判断样品中存在吗啡。质谱分析参数见表2。
在5.3.1和5.3.2的仪器条件下,吗啡的参考保留时间为2.25min。标准溶液的液相色谱-高分辨质谱法色谱图和质谱图参见图1、图2和图3。
5.3.4定量测定
5.3.4.1标准曲线的绘制:将标准系列工作液分别注入仪器(3.1)中,测定相应的响应值,以标准工作液的浓度为横坐标,以吗啡的母离子响应值为纵坐标,绘制标准曲线。
5.3.4.2试样溶液的测定:将试样溶液注入仪器(3.1)中,测定相应的响应值,根据标准曲线得到试样溶液中吗啡的浓度,试样溶液中吗啡的母离子响应值应在仪器的检测线性范围内。对于高浓度试样溶液须适当进行稀释,外标法定量。
6.数据处理
按公式(1)计算试样中吗啡的含量:
式(1)中:
x——试样中吗啡含量,单位为微克每升(μg/l);
c——从标准曲线计算得到的试样的吗啡浓度,单位为微克每升(μg/l);
c0——从标准曲线计算得到的空白试样的吗啡浓度,单位为微克每升(μg/l);
vi——试样溶液最终定容体积,单位为毫升(ml);
v——最终试样溶液代表的样品体积,单位为毫升(ml);
注:按照gb/t8170-2008规定,计算结果保留两位有效数字。
7.精密度
7.1重复性:在同一实验室,由同一操作者使用相同设备,按相同的测试方法,并在短时间内对同一被测对象相互独立进行测试获得的两次独立测试结果的绝对差值不大于这两个测定结果的算术平均值的15%,以大于这两个测定结果的算术平均值的95%的情况不超过5%为前提。
7.2再现性:在不同实验室,由不同操作者使用不同设备,按相同的测试方法,对同一被测对象相互独立进行测试获得的两次独立测试结果的绝对差值不大于这两个测定结果的算术平均值的20%,以大于这两个测定结果的算术平均值的95%的情况不超过5%为前提。
8.测定低限:1.0μg/l。
以上是本专利方法的全过程,前处理方法简单,采用试剂和材料较少,测定低限是同类检测项目中最低的,是e-9级别,而液相色谱-高分辨质谱法和同类检测项目不同检测方法比较精度最高的,测量质量数能够精确到小数点后五位。
根据操作步骤5.1和5.2可知,该方法的前处理简便,使用试剂较少,无需衍生,大量地节省人力和经济成本。根据5.3可知,定量和定性方法操作性强,灵敏度、准确度和仪器精度更高,由8测定低限1.0μg/l可知,是目前所有检测方法中最低的,而且减少假阳性检出的风险。
9.线性关系
在已确定的液相色谱-高分辨质谱仪的检测条件下,对吗啡浓度为1.0μg/l、2.0μg/l、5.0μg/l、10.0μg/l、20.0μg/l的标准溶液测定,以浓度与峰面积进行线性回归分析,线性回归方程见表3,标准曲线见图4。
10.回收率和精密度
采用添加法,即对本底不含吗啡的尿液样品(由健康儿童志愿者提供),添加吗啡浓度为1.0µg/l、2.0µg/l、10.0µg/l标准溶液三个水平的样品进行回收测定,每水平单独测定6次,回收率和精密度试验结果详见表4和表5。
由表5吗啡在不同添加浓度的变异系数(cv)可知其数值。根据变异系数的要求,在进行数据统计分析时,如果变异系数大于15%,则考虑该数据离散程度高,不与采用。本表中添加浓度1.0μg/l的变异系数(cv)值最大,为4.05%,远远小于15%,因此可证明该方法的精密度很高,方法复现性强。
上面描述,只是本发明的具体实施方式,各种举例说明不对本发明的实质内容构成限制。
- 还没有人留言评论。精彩留言会获得点赞!