一种气相色谱测萃取残液中氯仿含量的方法与流程
本发明涉及一种气相色谱测萃取残液中氯仿含量的方法,属于分析检测技术领域。
背景技术:
氯仿是化工行业常用的萃取剂之一,芳纶1414的生产过程中所用的n-甲基吡咯烷酮、芳纶ⅲ的生产过程中所用的n,n-二甲基乙酰胺,都是利用氯仿作为萃取剂进行萃取回收。为配合生产过程中检测萃取塔进出口的萃取残液废水的氯仿含量,定量计算萃取效率,检测水体排放水平,无论是从提高经济效益,生态环境角度来看,实现对其含量的快速、准确检测都显得尤为重要。
该检测方法可准确、快速地分析萃取残液废水中的氯仿。对工业化生产及环境监测具有一定的实际意义。
目前多采用外标法进行水体中氯仿的检测,对进样准确性要求极高。寻找对进样准确性要求低,且测量结果准确的氯仿含量测量方法,是目前需要解决的问题。
技术实现要素:
本发明要解决的技术问题是如何克服现有技术的不足,提供一种气相色谱测萃取残液中氯仿含量的方法。该发明所述方法对操作者进样准确度要求不高,操作简便易行,能有效减少操作误差,实现快速,准确检测分析。
为了实现上述目的,本发明的技术方案如下:
本发明公开了四氯化碳作为内标物在测量萃取液中氯仿含量的应用。通过四氯化碳作为内标物,制备内标物溶液,采用内标法测定萃取液中氯仿的含量。
本发明中,测量萃取液中氯仿含量的范围为50mg/l~5000mg/l。
本发明公开了一种气相色谱测萃取残液中氯仿含量的方法,包括以下步骤:
a.萃取
量取水样于分液漏斗中,加萃取剂振荡,振荡至少10分钟;静置分层,静置至少10分钟;得到萃取液;所述水样与萃取剂的体积比为0.5~10。
b.制备样品溶液
取步骤a萃取液的萃取剂层,加入内标物溶液,得到样品溶液;所述内标物溶液以四氯化碳作为内标物;所述内标物溶液的浓度为20g/l~40g/l;加入的内标物溶液与萃取剂层的体积比应确保氯仿与内标物的峰面积之比在0.1~10之间。
c.气相色谱分析
取步骤b得到的样品溶液进行气相色谱检测,获得色谱分析数据。
d.计算
将步骤c得到的色谱分析数据与预定氯仿标准数据比对,计算得到氯仿的含量;所述预定氯仿标准数据为用加有内标物溶液的氯仿系列标准溶液,在设定的气相色谱条件下测得的氯仿的校正因子。
进一步地,本发明在步骤a中,使用的萃取剂为正戊烷或者正庚烷。
进一步地,本发明在步骤b中,所述内标物溶液的制备方法为:称量2g~4g四氯化碳,用正戊烷或者正庚烷定容于100ml容量瓶中,摇匀。
进一步地,本发明在步骤c中,气相色谱分析的毛细管色谱柱采用中等极性或极性毛细管色谱柱。
进一步地,本发明在步骤c中,气相色谱检测采用的是fid检测器。
进一步地,本发明在步骤c中,氮气为载气,流量为30ml/min,氮气:氢气:空气流量之比为1:1:10。
本发明在步骤c中,进样温度为210~250℃。确保从水样萃取到的n,n甲基吡咯烷酮,n,n-二甲基乙酰胺等。
本发明在步骤c中,柱温采用程序升温起始,柱温50℃,保持3分钟后,以20℃/min的升温速率至180℃保持5min组分也能瞬间汽化载入色谱柱而有效分离。
本发明在步骤c中,柱流速为1.0~2.0ml/min。该柱流速在上述进样温度和程序升温条件能得各组分的较好分离。
进一步地,本发明在步骤c中,fid检测器的温度为240~280℃。
本发明带来的有益效果为:
(一)本发明采用了气相色谱内标法。以中等极性或极性的毛细管色谱柱作为分离柱,峰形尖锐,分离度好。内标法对操作者进样准确度要求不高,操作简便易行,能有效减少操作误差,实现快速,准确检测分析。本方法优选到四氯化碳作为内标物,以内标法定量,有效减少了因进样量、溶剂损失、待测样品的浓度等各种因素造成的误差,即使测定条件略有变化也不会对结果产生影响,相对于外标法定量,对仪器用具和人员操作的要求都不高。该方法操作简便,利用配备常规检测器与用具,就能获得较为理想的测定结果。该方法准确度和精密度良好,是一种简便、可行的测定萃取残液废水中微量氯仿的方法。
(二)本发明的测量结果中,标准曲线的相关系数可达0.9997,接近于1;平均回收率在96.1%~100.0%;系统适用性好,氯仿、内标物四氯化碳分离度r能达2,理论塔板数大于5000,能满足实验要求,具有广泛适用性。
(三)本发明的专属性强,氯仿、四氯化碳、正戊烷与在芳纶1414、芳纶ⅲ的生产中产生的萃取残液里的n-甲基吡咯烷酮、n,n-二甲基乙酰胺在本发明的色谱条件下也能完全分离,且空白不干扰测定。
附图说明
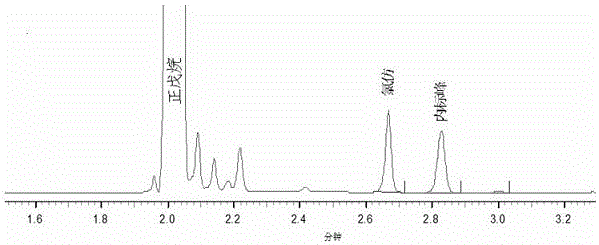
图1为正戊烷、氯仿和四氯化碳标准溶液谱图。
图2为线性关系图。
图3为实施例9样品溶液色谱图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全面的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
实施例1
本发明公开了以四氯化碳作为内标物的气相色谱内标法测量萃取液中氯仿含量的应用。通过四氯化碳作为内标物,制备内标物溶液,采用内标法测定萃取液中氯仿的含量。
本发明中,测量萃取液中氯仿含量的范围为50mg/l~5000mg/l。
实施例2
本实施例公开了一种气相色谱测萃取残液中氯仿含量的方法,包括以下步骤:
a.内标溶液的配制
称量2g~4g四氯化碳,用正戊烷定容于100ml容量瓶中,摇匀。
b.系列标准溶液的配制
称取约0.5g~1g氯仿,用正戊烷定容于100ml容量瓶中,摇匀,作为标准母液。再取不同体积的标准母液,加正戊烷配制成等体积的含氯仿10mg~50mg不同浓度的系列标准溶液,再加等体积的内标溶液。氯仿与内标物的峰面积之比在0.1~10之间。
c.样品溶液的制备
量取一定体积水样于分液漏斗中,加萃取剂正戊烷至少10ml;充分振荡进行萃取,振荡至少10钟;静置分层,静置至少10分钟;上层为萃取液;所述水样与萃取剂的体积比为0.5~10。
准确移取萃取液,移取体积与步骤b中未加内标物的标准溶液相同,但不超过萃取时加入萃取剂的体积量,加入与配制标准溶液时等体积的内标物溶液,得到样品溶液。
d.测定校正因子
用步骤b中的系列标准溶液,分别在设定的气相色谱条件下进样得到色谱数据,以峰面积比和含量比绘制标准曲线,测得校正因子。
e.气相色谱分析
取步骤c得到的样品溶液进行气相色谱检测,获得色谱分析数据,用步骤d测得的校正因子计算得到氯仿的含量。
实施例3
本实施例在实施例2的基础上作进一步改进:萃取剂正戊烷可替换为正庚烷。
实施例4
本实施例在实施例2的基础上作进一步优化:在步骤d中,气相色谱分析条件毛细管色谱柱采用中等极性或极性毛细管色谱柱,如db-35,peg-20m等。
实施例5
本实施例在实施例2的基础上作进一步优化:步骤d中,气相色谱分析条件进样温度为220℃。
实施例6
本实施例在实施例2的基础上作进一步优化:在步骤d中,气相色谱分析条件柱流速为1.5ml/min。
实施例7
本实施例在实施例2的基础上作进一步优化:在步骤d中,气相色谱分析采用的是fid检测器,fid检测器的温度为250℃。
实施例8
本实施例在实施例2的基础上作进一步优化:在步骤d中,载气为氮气,流量为30ml/min,氮气:氢气:空气流量之比为1:1:10。
实施例9
本实施例测定含有n-甲基吡咯烷酮的萃取残液废水中的微量氯仿含量
a.色谱条件
本发明采用agilent7820a气相色谱仪,氮气钢瓶中氮气纯度为99.999%,氢气钢瓶中氢气纯度为99.999%,空气为钢瓶干燥压缩空气。使用以下色谱条件:
检测器为fid检测器,检测器温度250℃;色谱柱为db-35毛细管色谱柱,规格为30m×0.32mm×0.25μm;进样温度为220℃;柱温采用程序升温起始柱温50℃,保持3分钟后,以20℃/min的升温速率至180℃保持5min;氮气为载气,流量为30ml/min,氮气:氢气:空气流量之比为1:1:10,柱流速为1.5ml/min;进样体积为1μl;分流比为100:1。
b.溶液配制
(1)内标物溶液的配制
称量2.3302g四氯化碳,以正戊烷定容于100ml容量瓶中,摇匀备用。
(2)标准母液的制备
称量0.5011g氯仿,以正戊烷定容于100ml容量瓶中,摇匀备用,其中氯仿含量为5.011g/l。
(3)系列标准溶液的配制
表1:系列标准溶液的配制
(4)样品溶液的制备
取萃取残液废水100.0ml于分液漏斗中,再加20.00ml正戊烷,充分振荡萃取残液废水的氯仿,取上层萃取液10.00ml,加2.00ml内标物溶液,充分摇匀,制得样品溶液。
c.气相色谱分析
在上述色谱条件下,5个不同的标准溶液,每个标准溶液分别进样2次,通过以下公式(1)计算校正因子,以峰面积比和含量比绘制标准曲线,建立相应的内标分析方法,线性关系图如图2。
式中:as——内标物质的峰面积;
ar——标准品的峰面积;
cs——内标物质的浓度;
cr——标准品的浓度。
表2:系列标准溶液色谱数据统计表
系列标准溶液色谱数据统计如表2,计算平均校正因子f为0.029448,其相对标准偏差为1.69%,不大于2%,相关系数为0.9994,符合实验要求。
在与标准溶液相同的色谱条件下,选取相应的内标分析方法,制备两个平行样品溶液进样测定,每个样品溶液进样2次,色谱谱图如图3,数据记录如表3,通过公式(2)计算氯仿含量。
式中:
ax——样品溶液的峰面积;
cx——萃取残液废水的氯仿含量,单位为毫克每升(mg/l);
as’——内标物质的峰面积;
cs——内标物质的浓度;
f——校正因子。
表3:样品溶液测定记录
取平均值为该萃取残液废水中的氯仿含量,即为3410mg/l,极差为156mg/l,rsd为1.91%。
实施例10
分别配制3个氯仿含量不同的标准水溶液,再分别萃取、制样、色谱测定,其记录结果如表4:
表4:测定水中氯仿含量的精密度和准确度数据统计表
由结果可见,本发明涉及的氯仿含量测定方法精密度和准确度良好,平行结果相对标准偏差、测定结果平均值与标准值的相对误差均小于5%,能满足一般实验要求。
以上所述,仅是本发明的较佳实施例,并非对本发明做任何形式上的限制,凡是依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化,均落入本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!