鉴定蛋白质相互作用的方法与流程
相关申请的交叉引用
本申请要求2017年8月18日提交的新加坡临时申请第10201706788w号的优先权权益,其内容出于所有目的通过引用整体并入本文。
本发明一般涉及分子生物学和计算生物学领域。特别地,本发明涉及直接在细胞和组织中鉴定和监测蛋白质-蛋白质相互作用的方法。
背景技术:
活细胞源自在时间和空间上,在蛋白质与核酸、代谢物和脂质之间发生的无数生物分子相互作用。这种复杂的生物网络的中心是介导细胞的生物化学过程和结构组织的蛋白质复合物。它们根据细胞需要动态装配和解离,并且富含牵涉遗传和感染疾病的蛋白质。
蛋白质可以在不同的物理、细胞或生理状态和条件下彼此相互作用以形成蛋白质复合物。这些相互作用是复杂的和动态的,其中蛋白质和蛋白质复合物可以根据细胞的需要进行装配或解离。至关重要的是,能够进行实时观察并在蛋白质组水平(proteome-widelevel)上研究细胞内的这些相互作用以便更好地理解生理状态中的这些复杂相互作用。迫切需要这样的方法,因为它们将缓和当前在理解动态蛋白质相互作用的复杂性方面的分歧,这些相互作用可用于鉴定在疾病中被调节的蛋白质复合物。
已经进行了大规模的努力以使用特定的细胞系来填充合理的人类蛋白质复合物库。来自这些工作和其它工作的数据,加上专心致志的努力,已经积聚成一个大型蛋白质-蛋白质相互作用网络,该网络描述了人类蛋白质组的合理的细胞网络(cellularwiring)和功能组织。然而,各种细胞类型、生理状态和疾病状况下装配的蛋白质网络以及鉴定的蛋白质复合物的保守性尚不清楚。
需要提供允许直接在细胞和组织中对蛋白质-蛋白质相互作用和蛋白质复合物的形成和动力学进行有效、全系统监测和无假设(hypothesis-free)鉴定的方法。
技术实现要素:
在一个方面,本发明涉及一种用于鉴定一种或多种第一蛋白质与一种或多种其它蛋白质之间的蛋白质相互作用的方法,所述方法包括以下步骤:
a)使包含一种或多种第一蛋白质和所述一种或多种其它蛋白质的一种或多种样品暴露于至少一种预选条件,达至少一个预选的持续时间;
b)将所述一种或多种样品的不溶性级分(insolublefraction)与至少一种可溶性级分(solublefraction)分离并分开;以及
c)分析步骤b)的至少一种可溶性级分或不溶性级分,以鉴定所述一种或多种样品中一种或多种第一蛋白质与一种或多种其它蛋白质之间的所述蛋白质相互作用。
在另一方面,本发明涉及本文所述的方法用于筛选治疗药物靶标、疾病进展预后、测定药物治疗的功效、测定蛋白质化学计量、鉴定新的蛋白质途径、绘制蛋白质相互作用组(proteininteractome)图谱、预测蛋白质功能或绘制时阈蛋白质相互作用(temporalproteininteraction)图谱的用途。
定义
本文所用的以下词语和术语应具有所指示的含义:
“蛋白质”是指氨基酸残基的聚合物,其中蛋白质可以是单分子或可以是多分子复合物。本文所用的术语可以指任何大小、结构或功能的多分子复合物、多肽、肽、寡肽中的亚基。通常理解,肽的长度可以是2至100个氨基酸,而多肽的长度可以超过100个氨基酸。蛋白质也可以是天然存在的蛋白质或肽的片段。术语蛋白质还可以应用于氨基酸聚合物,其中一个或多个氨基酸残基是相应天然存在的氨基酸的人工化学类似物。蛋白质可以是野生型的、重组的、天然存在的或合成的,并且可以构成天然存在的或非天然存在的多肽的全部或部分。蛋白质复合物的亚基和蛋白质可以相同或不同。蛋白质也可以是功能性的或非功能性的。
“蛋白质相互作用(proteininteraction)”或“蛋白质-蛋白质相互作用(protein-proteininteraction)”是指两种或更多种蛋白质之间的缔合。蛋白质-蛋白质相互作用可影响单个蛋白质的性质或功能的改变。蛋白质相互作用也可以在细胞或生物体中引起生物学效应。蛋白质相互作用可以是稳定的或短暂的。这些相互作用中的每一种都可以是强的或弱的。通常理解的是,蛋白质相互作用的强度和特异性受性质影响,所述性质例如但不限于蛋白质结构、蛋白质结构域、蛋白质浓度、热稳定性、溶解度、温度、质量、ph、密度、体积、分子键、电荷、化学稳定性、粘度、熔点、沸点或其任意组合。较强的蛋白质相互作用可以通过共价键如二硫键或电子共享来实现,而较弱的蛋白质相互作用可以通过非共价键如氢键、范德华力、疏水键或离子相互作用来建立。
“样品”是指取自、获自或来源于生物体的样本。样品可以获自或来源于任何多细胞或单细胞生物,例如但不限于动物、植物、真菌、细菌、古细菌、病毒或原生生物。
“沉淀”是指相互作用的蛋白质在变性时的共聚集。可以通过一种或多种方式,例如热、化学、辐射、ph、超声处理或其任意组合来诱导变性。
蛋白质在变性时解折叠(unfold)并聚集或沉淀。相互作用的蛋白质在变性时可以共聚集。聚集或沉淀的蛋白质是不溶的,在不溶级分中是可检测的,而在可溶级分中是不可检测的。当以不同的持续时间暴露于多种条件时,不同的蛋白质以不同的条件聚集。可以在蛋白质以不同的持续时间暴露于不同条件时测量聚集或保持可溶的蛋白质的百分比,并且可以以曲线形式表示在一定条件范围的蛋白质的溶解度。
如本文所用,蛋白质的“溶解度(solubility)”是指蛋白质在不同条件下的溶解度。蛋白质在变性时解折叠并聚集或沉淀。相互作用的蛋白质在变性时可以共聚集。可以在蛋白质以不同的持续时间暴露于不同条件时测量聚集或保持可溶的蛋白质的百分比,并且可以以溶解度曲线形式表示在一定条件范围的蛋白质的溶解度。例如,当以不同的持续时间暴露于一定的温度范围时,不同的蛋白质在不同的温度下聚集。可以在蛋白质以不同的持续时间暴露于不同温度时(热溶解度(ts))时测量聚集或保持可溶的蛋白质的百分比,并且可以以熔解曲线的形式表示在一定温度范围的蛋白质的热溶解度。
如本文所用,“溶解度曲线(solubilitycurve)”是指对蛋白质解折叠和共聚集事件的分析或评估。术语“熔解曲线(meltcurve或meltingcurve)”是指当利用热使蛋白质变性时对蛋白质解折叠和共聚集事件的分析或评估。
“邻近共聚集(proximityco-aggregation,pca)”是对在不同条件下变性时蛋白质共聚集的量度。相互作用的蛋白质在变性时共聚集,导致不同条件下相似的溶解度。pca可用于全系统细胞内研究和蛋白质复合物动力学的监测,其中相互作用的蛋白质在变性时共聚集,导致不同条件下的相似溶解度。
“邻近共聚集(pca)特征(signature)”是指使用pca鉴定的蛋白质相互作用的模式(pattern)。pca特征与相互作用化学计量和丰度化学计量正相关,其中如本文所用的术语“相互作用化学计量(interactionstoichiometry)”是指蛋白质之间的最佳相互作用程度,并且“丰度化学计量(abundancestoichiometry)”是指相互作用蛋白质的丰度之间的比率。
“距离度量(distancemetric)”是指两个数据点之间的距离的测量。距离度量可用于测量两条溶解度曲线或熔解曲线之间的相似度(similarity)。
附图说明
当结合非限制性实施例和附图考虑时,通过参考具体实施方式将更好地理解本发明,在所述附图中:
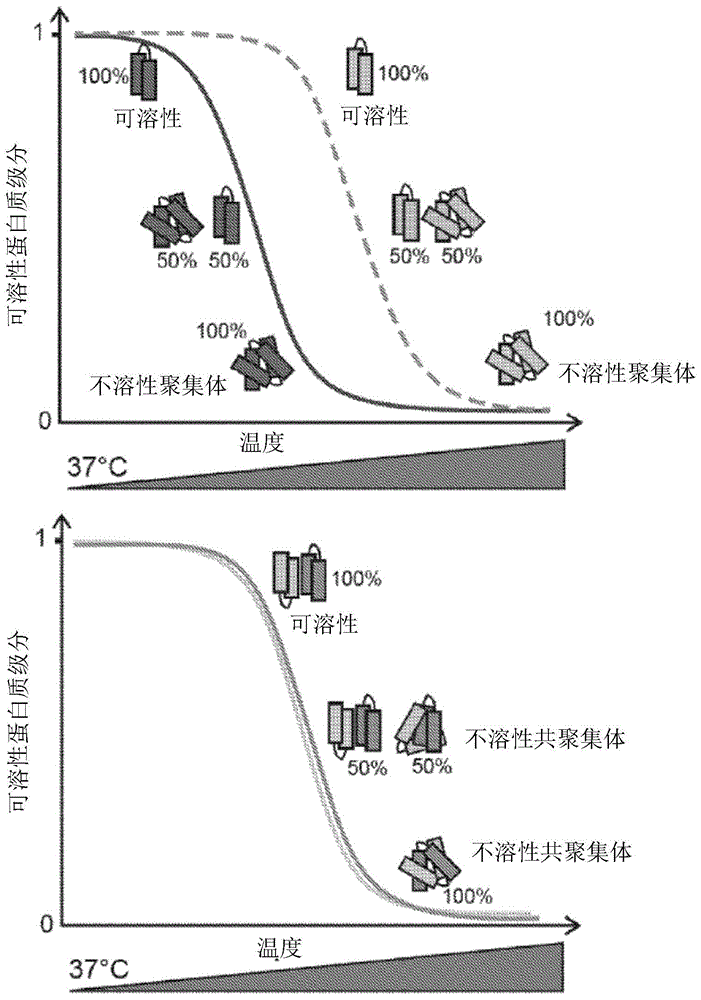
图1显示了表示用于监测蛋白质-蛋白质相互作用的pca原理的示意图:相互作用的蛋白质之间的共聚集和沉淀导致在不同变性温度下相似的蛋白质溶解度。该图表示在不同温度点的非相互作用蛋白质(左)和相互作用蛋白质(右)的可溶性蛋白质的级分。
图2显示了代表cdk2-ha、细胞周期蛋白e1-v5、cdk2-ha(+细胞周期蛋白e1-v5)和细胞周期蛋白e1-v5(+cdk2-ha)的熔解曲线的散点图。共同相互作用(cointeracting)的细胞周期蛋白e1和cdk2表现出相似的熔解曲线:hek293t细胞用编码带有v5标签的细胞周期蛋白e1和/或带有ha标签的cdk2的质粒转染,随后进行完整的细胞cetsa实验。数据来自三个生物学重复的免疫印迹(平均值±sd)并用三参数对数逻辑函数拟合。
图3显示的示意图显示ms-cetsa实验的概况。m/z,质/荷比;tmt,串联质谱标签。
图4显示了2个水平箱形图,其表示熔解曲线相似度的分布(基于蛋白质对之间的欧式距离(euclideandistance))。(a)显示根据一些报道出版物的已知相互作用蛋白质对之间的熔解曲线相似度的分布:pmid,pubmed标识符。深灰色点表示每个相应蛋白质子集的全部蛋白质对的中值。(b)显示的水平箱形图表示已知相互作用蛋白质对和corum中相互作用蛋白质对的熔解曲线相似度的分布。
图5显示了9个s形曲线图(sigmoidgraphs),它们代表所选择的蛋白质复合物的熔解曲线:浓缩蛋白(condensin)复合物(约4000种其它蛋白质的熔解曲线以灰色绘制)、20s蛋白酶体、escrt-ii复合物、arp2/3蛋白复合物、baf复合物、后期促进(anaphase-promoting)复合物、bloc-1(溶酶体相关细胞器复合物1的生物发生)、保守寡聚高尔基体(cog)复合物和多合成酶复合物。黑线代表复合亚基。
图6显示了2个s形曲线图,它们代表所有定量蛋白质的熔解曲线的中值(左)和pa700复合物亚基的熔解曲线的中值(右),其中细胞裂解物已经进行了脱盐。
图7显示了一个水平箱形图,它代表由细胞裂解物和完整细胞数据计算的已知相互作用蛋白质对的熔解曲线相似度(基于蛋白质对之间的欧式距离)的分布。深灰色点指示每个相应蛋白质子集的全部蛋白质对的中值。
图8显示了散点图,它代表在由来自细胞裂解物和完整细胞的蛋白质计算的不同统计阈值下具有非随机pca行为的corum蛋白质复合物的数量。
图9显示了4个s形曲线图,它们代表细胞裂解物和完整细胞中dna-染色质以及膜相关复合物nup107-160亚复合物和起始识别复合物(originrecognitioncomplex)的亚基的熔解曲线。
图10显示了6个s形曲线图,它们代表细胞和裂解物不同的亚复合物的实例的熔解曲线,例如细胞裂解物和完整细胞中的(a)brd4-rfc复合物、ndc80着丝粒,以及(b)40s核糖体亚基和60s核糖体亚基。
图11显示了ndc80着丝粒的结构示意图。
图12显示了4个箱形图,它们代表pca特征(完整细胞ms-cetsa数据)与(a)蛋白质相互作用化学计量、(b)已知相互作用蛋白质对的蛋白质丰度化学计量的相关性的数据。(c)显示了在两个参数中均具有高化学计量的蛋白质对。(d)显示了相互作用和丰度化学计量的乘积与表现出最高pca特征的pca特征更好地相关。
图13显示了代表蛋白质复合物的功能组织的核心附着模型(coreattachmentmodel)的示意图。
图14显示了代表数据的点阵图,所述数据显示复合物的核心-附着蛋白质对通常表现出比核心-核心蛋白质对更弱的pca特征。每个点代表蛋白质复合物。
图15显示了8个s形曲线图,它们代表来自不同细胞条件下的细胞的染色质装配复合物(caf-1)、染色质重塑baf复合物、tnfα/nfκb信号转导复合物和trex/tho复合物的亚基的熔解曲线。
图16显示了蛋白质印迹分析的照片。(a)显示了在以下样品(c:对照(dmso);t:tnfα;m:甲氨蝶呤;t+m:tnf-α+甲氨蝶呤)中,使用(p-ikkα/β(s180/181)、p-ikbα(s32)、ikbα、p-p65和(s536)评估nfκb信号转导途径成员。hsp90用作上样对照。(b)显示了样品c、t、m和t+m中nfκbp65和p50的免疫沉淀分析。观察到ikbα、p65、p50水平,并将hsp90用作上样对照。
图17显示了与未同步化的细胞相比,在细胞周期s期捕获的细胞中pca行为具有非随机性差异的蛋白质复合物的网络示意图。
图18显示了柱状图,其代表表现出具有p值<0.05和p<0.01的pca复合物的蛋白质复合物的数量。
图19显示了4个s形曲线图,它们代表具有强pca特征(p值<0.001)的所选复合物的熔解曲线。复合物是从小鼠肝组织获得的cct复合物(含有伴侣蛋白的tcp1复合物)、黏连蛋白-sa2复合物、arp2/3蛋白复合物和pa700复合物。
图20显示的示意图代表6种不同细胞系hct116、a375、mcf7、hek293t、k562和hl60中raf-mek-erk途径中差异相互作用的pca分析报告(基于pca导出的z分数)。pca导出的z分数用于帮助各细胞系间的pca特征的比较。较高的z分数意味着增加的相互作用。
图21显示了6个s形曲线图,它们代表6种不同细胞系hct116、a375、mcf7、hek293t、k562和hl60中raf1和braf1的熔解曲线。
图22显示了2个散点图,它们代表用于在蛋白质相互作用网络上使用图形或网络聚类算法预测corum蛋白质复合物的数据,所述蛋白质相互作用网络通过出版物计数(publicationcount)、可靠性分数(出版物计数+功能相似度)和基于pca的评分进行加权。精确度和召回率(recall)针对corum参照复合物(大小>3)进行评估,其中jaccard相似度≥0.5认为匹配。
图23显示了代表接收者操作特征曲线(receiveroperatingcharacteristiccurve,roc)的2个曲线图。(a)显示了代表使用pca的已知相互作用蛋白质对的可预测性的数据。(b)显示了代表使用pca的共复合物(co-complex)对的可预测性的数据。使用完整k562细胞的熔解曲线数据。计算具有熔解曲线的全部蛋白质对之间的欧氏距离并按升序排列(较低的欧氏距离优先)。除了已知相互作用或被注释为corum复合物一部分的那些,全部蛋白质对均被视为阴性数据。
图24显示了蛋白质印迹分析的照片。(a)显示了细胞周期蛋白e1-v5和cdk2-ha的共免疫沉淀的评估。(b)显示了细胞周期蛋白e1-v5和cdk2-ha在不存在和存在其相互作用配偶体(partner)的情况下在不同温度下的溶解度。进行三个生物学重复,并显示了代表性的印迹。
图25显示了水平箱形图,它代表了每个过滤后的相互作用数据集中蛋白质之间曲线相似度的零分布(nulldistribution)。null1、null2、null3和null4是分别由pmid≥1、pmid≥2、pmid≥3和pmid≥4报告的相互作用数据中的全部蛋白质产生的曲线相似度分布。
图26显示了3个散点图,它们代表了不同样品上每种蛋白质复合物的亚基之间的平均欧式距离。(a)显示了脱盐细胞裂解物对蛋白质复合物的平均熔解曲线相似度的影响。对原始样品和脱盐样品进行cetsa实验。在计算中仅使用在两个数据集中都存在的蛋白质。(b)显示了来自两个不同批次的k562细胞的两次裂解物cetsa实验中针对每种蛋白质复合物计算的平均欧式距离的再现性。(c)显示了来自两个不同批次的k562细胞的两次裂解物cetsa实验中针对每种蛋白质复合物计算的平均欧式距离的再现性,但是仅考虑具有至少三个具有定量信息的肽的蛋白质且其不必唯一。
图27显示了6个s形曲线图,它们代表视觉鉴定的蛋白质复合物子集的熔解曲线,所述蛋白质复合物在其亚基的熔解曲线之间具有非随机相似度,但似乎由于作为整个复合物脱盐而热不稳定。所鉴定的蛋白质复合物子集是浓缩蛋白i复合物、mcm复合物、多合成酶复合物、pa700复合物、40s核糖体亚基(细胞质)和ranbpm-muskelin-twa1-hsmpp8复合物。
图28显示了2个散点图和2个箱形图,它们代表用于比较完整细胞和裂解物cetsa数据的蛋白质复合物的平均欧式距离。(a)显示了针对每种蛋白质复合物计算的亚基之间的平均欧式距离的散点图。(b)显示了针对每种蛋白质复合物计算的亚基之间平均欧式距离分布的箱形图。(c)显示了针对在完整细胞或裂解物cetsa数据中具有非随机pca特征的蛋白质复合物子集计算的亚基之间的平均欧式距离的散点图。(d)显示了针对在完整细胞或裂解物cetsa数据中具有非随机pca特征的蛋白质复合物子集计算的亚基之间的平均欧式距离的箱形图。
图29显示了代表在完整细胞和裂解物cetsa数据中具有非随机pca特征的蛋白质复合物的维恩图(venndiagram)。
图30显示了代表细胞和裂解物中总蛋白质复合物的级分的散点图。一部分合格(qualified)蛋白质复合物在单个裂解物和细胞数据集中具有非随机pca行为。统计学阈值设定为p值<0.05。每个节点表示数据集;红色水平线表示每种数据类型内的平均值,垂直线表示平均值的标准误差。
图31显示了在不同类别、应激反应、细胞周期和dna加工下,细胞和裂解物中每个功能分类中pca显著的蛋白质复合物的百分比的柱状图。在三种corum功能类别下的蛋白质复合物的数量已被鉴定为在统计学上富含完整细胞或裂解物数据中pca显著的蛋白质复合物。
图32显示了欧式距离与不同参数的3个散点图。(a)显示了欧式距离与相互作用化学计量。(b)显示了欧式距离与丰度化学计量。(c)显示了相互作用的蛋白质对的欧式距离与其乘积值。
图33显示了代表每组中全部蛋白质对的熔解曲线之间的欧式距离的3个箱形图,其中具有相似丰度的蛋白质对不显示强pca特征。(a)代表按照光谱计数首先对蛋白质进行分类,然后分成每组400个蛋白质的组。计算每组中每种可能的蛋白质配对组合的熔解曲线(完整的k562数据)之间的欧式距离,并通过箱形图使分布可视化。(b)代表对蛋白质的相似分析,将得到最可重现的曲线(定义为定量psm>10的曲线)。(c)代表与(a)和(b)相似的分析,但是使用用蛋白质组学标尺方法估算的绝对蛋白质丰度。
图34显示的图代表用碘化丙啶染色的facs分析对甲氨蝶呤处理和dmso处理的k562的dna含量分析。
图35显示散点图,它代表dmso处理和甲氨蝶呤处理的k562细胞之间的相对蛋白质表达在两个生物学重复中的再现性。用于生成实验用熔解曲线的37℃样品用tmt试剂标记并通过ms进行分析。
图36显示了针对多个细胞系生成的ms-cetsa数据的2个柱状图。(a)代表每个绘制的细胞系中鉴定的蛋白质数量。(b)代表每个绘制的细胞系中具有非随机pca特征的合格corum复合物(具有熔解曲线的3个或更多个亚基)的百分比。
图37显示了在6个细胞系的任一个中具有强pca特征(p<0.001)的蛋白质复合物的pca特征的热图。亚基的比例是指其中熔解曲线在我们的ms-cetsa实验中被定量的亚基部分。每种复合物的第一个数字是corum复合物的标识号。
图38显示了代表各生物学重复间的相同蛋白质的熔解曲线之间的欧式距离的箱形图,其中曲线再现性与定量psm的数量相关。
图39显示了12个s形曲线图,它们代表所绘制的6个细胞系中的前折叠素复合物和eif3核心复合物的亚基的熔解曲线。
具体实施方式
在第一方面,本发明涉及一种用于鉴定一种或多种第一蛋白质与一种或多种其它蛋白质之间的蛋白质相互作用的方法,所述方法包括以下步骤:
a)使包含一种或多种第一蛋白质和所述一种或多种其它蛋白质的一种或多种样品以至少一个预选的持续时间暴露于至少一种预选条件;
b)将所述一种或多种样品的不溶性级分与至少一种可溶性级分分离并分开;以及
c)分析步骤b)的至少一种可溶性级分或不溶性级分,以鉴定所述一种或多种样品中一种或多种第一蛋白质与一种或多种其它蛋白质之间的所述蛋白质相互作用。
一种包含一种或多种第一蛋白质和一种或多种其它蛋白质的样品可以暴露于至少一种预选条件。与不和一种或多种其它蛋白质相互作用时的第一蛋白质相比,该预选条件能够引起和一种或多种其它蛋白质相互作用时的第一蛋白质沉淀的变化。在一个实例中,沉淀的变化可以是沉淀的增加或减少。在另一个实例中,与不和一种或多种其它蛋白质相互作用时的第一蛋白质相比,该预选条件能够引起和一种或多种其它蛋白质相互作用时的第一蛋白质沉淀。
在一个实施方案中,步骤a)中的至少一种预选条件可以是温度、ph、浓度、化学品(例如尿素)、辐射(例如微波)、超声处理或其任意组合。在优选实施方案中,预选条件是温度或温度范围。通常应当理解,在本发明中可以使用一定范围的温度。例如,温度可以为约30℃至约70℃。例如,约31℃至约69℃、约32℃至约68℃、约33℃至约67℃、约34℃至约66℃、约35℃至约65℃、约36℃至约64℃、约37℃至约63℃、约38℃至约62℃、约39℃至约61℃、约40℃至约60℃、约41℃至约59℃、约42℃至约58℃、约43℃至约57℃、约44℃至约56℃、约45℃至约55℃、约46℃至约54℃、约47℃至约53℃、约48℃至约52℃或约49℃至约51℃。在另一个实施方案中,温度可以为约37℃至约64℃。在另一个实施方案中,预选条件包括约37.0℃、约38.0℃、约39.0℃、约40.0℃、约41.0℃、约42.0℃、约43.0℃、约44.0℃、约45.0℃、约46.0℃、约47.0℃、约48.0℃、约49.0℃、约50.0℃、约51.0℃、约52.0℃、约53.0℃、约54.0℃、约55.0℃、约56.0℃、约57.0℃、约58.0℃、约59.0℃、约60.0℃、约61.0℃、约62.0℃、约63.0℃和约64.0℃的预选温度。在一个实例中,预选条件为37.0℃、40.0℃、43.0℃、46.0℃、49.0℃、52.0℃、55.0℃、58.0℃、61.0℃和64.0℃的预选温度,例如如图3所示。
包含一种或多种第一蛋白质和一种或多种其它蛋白质的样品可以以预选的持续时间暴露于至少一种预选条件。预选的持续时间可以以秒(s)、分钟(min)、小时(h)或天计。例如,预选的持续时间可以为10s至2min、1min至3min、2min至4min、3min至5min、4min至6min、5min至7min、6min至8min、7min至9min或8min至10min。在另一个实例中,预选的持续时间可以为1min、2min、3min、4min、5min、6min、7min、8min、9min或10min。在另一个实例中,预选的持续时间为3min。
在一个实施方案中,样品可以是生物样品。例如,样品可以是但不限于全血或其组分(例如血浆、血清)、尿、唾液淋巴、胆汁、痰、泪、脑脊髓液、支气管肺泡灌洗液、滑液、精液、腹水肿瘤液、乳汁、脓、羊水、口腔粘膜涂片、培养的细胞、从培养的细胞收集的培养基、细胞团块、从整个生物体或其组织、细胞或组分部分的一部分制备的裂解物、匀浆或提取物、或其级分或部分。在一个实例中,样品可以是从来自生物体的器官分离的细胞,其中器官可以是但不限于肝、脑、心脏、脾、肾、骨、淋巴结、肌肉、血管、骨髓、胰腺、肠、膀胱或皮肤。
在另一个实施方案中,生物样品可以是来自细胞系的培养细胞,所述细胞系例如但不限于hct116、a375、mcf7、hek293t、k562、hl60、hek293ft、hek293、hela、sf9、chos、jurkat、saos-2或pc3。在另一个实施方案中,样品可以是完整细胞或细胞裂解物。在优选的实施方案中,样品是完整细胞。
在一个实施方案中,一种或多种样品可从相同样品或不同样品中获得。例如,一种或多种样品可以是相同细胞系或细胞类型的一种或多种完整细胞或细胞裂解物。在另一个实例中,一种或多种样品可以是来自两种或更多种不同细胞系或细胞类型的一种或多种完整细胞或细胞裂解物。
在另一个实施方案中,样品可以被修饰,其中修饰包括纯化、稀释、改变ph、添加盐或除去盐、添加添加剂、添加药物或导向药物(druglead)、氧化或还原、暴露于化学品或化合物、改变细胞状态或生理状态、或改变细胞环境。在一个实例中,可以使用但不限于质粒或核酸通过转染来修饰样品,其中质粒包含编码标签的序列,所述标签例如但不限于gfp、ha、flag、mcherry、v5、gst、生物素、磷酸盐、组氨酸、链霉亲和素或其任意组合。在另一个实例中,可以通过进行脱盐、缓冲液交换、凝胶过滤层析、尺寸排阻层析或其任意组合来修饰样品。在另一个实例中,细胞裂解物如k562细胞裂解物可以脱盐以消耗低分子量(lmw)配体,从而一起干扰复合物并获得用于pca验证的ms-cetsa数据。还可以通过暴露于化学品来修饰样品,所述化学品包括但不限于甲氨蝶呤、二甲基亚砜(dmso)或肿瘤坏死因子α(tnfα)、或其任意组合。
第一蛋白质和/或其它蛋白质可以是相互作用的蛋白质、蛋白质复合物中的蛋白质、或蛋白质复合物中的亚基。在一个优选的实施方案中,第一蛋白质和/或其它蛋白质可以是野生型蛋白质或重组蛋白质。在一个实例中,所述第一蛋白质为带有v5标签的细胞周期蛋白e1,其通过与v5标签抗体预偶联的蛋白a琼脂糖珠进行鉴定和检测,所述其它蛋白质为cdk2。在另一个实例中,所述第一蛋白质可以为40s或60s核糖体亚复合物,所述其它蛋白质可以为40s或60s核糖体亚复合物,其中本领域通常已知40s和60s核糖体亚复合物形成核糖体复合物。
第一蛋白质和其它蛋白质可以来自相同或不同的样品。第一蛋白质和其它蛋白质也可以来自样品的相同或不同部分。在一个实例中,第一蛋白质和其它蛋白质可位于细胞裂解物内。在另一个实例中,第一蛋白质和其它蛋白质可以位于细胞内或细胞上。
所述一种或多种样品可以包含可溶性级分和不溶性级分。蛋白质在变性时解折叠和聚集或沉淀并且是不溶的。聚集的蛋白质在不溶性级分中是可检测的。未解折叠或天然形式的蛋白质在可溶性级分中是可检测的。
来自步骤a)的一种或多种样品的可溶性级分可以与不溶性级分分离并分开。在从样品中分离并分开级分的步骤之前,可以使用例如但不限于冻融、机械剪切、超声处理、离心、过滤或其任意组合的方法处理样品。然后可通过本领域已知的常规方法(实例包括但不限于吸移可溶性级分、倒出可溶性级分或其任意组合)实现可溶性级分与不溶性级分的分离并分开。
在一个实施方案中,在从不溶性级分中分离并分开至少一种可溶性级分之前,可以裂解样品或细胞以获得细胞裂解物。在另一个实施方案中,在从不溶性级分中分离并分开至少一种可溶性级分之前,可以将细胞裂解物离心或过滤以获得上清液级分。
在一些实施方案中,样品或细胞可以通过两个冻融循环和机械剪切被裂解,其中使用本领域通常已知的条件通过离心将上清液与细胞碎片和聚集的蛋白质团块(pellet)分离。
然后可以分析步骤b)的至少一种可溶性级分或不溶性级分,以鉴定样品中一种或多种第一蛋白质与一种或多种其它蛋白质之间的蛋白质相互作用。
在一个实例中,可以通过计算方法进行分析。在另一个实例中,可以通过包括计算算法的计算方法来进行分析。在又一个实例中,可以通过包括对数据去卷积(deconvolute)的计算算法的计算方法进行分析,以鉴定存在或被调节的蛋白质复合物或蛋白质-蛋白质相互作用。
在一个实施方案中,步骤c)中的分析可以进一步包括以下步骤:i)测定蛋白质的溶解度;以及ii)获得邻近共聚集(pca)特征以鉴定所述一种或多种样品中一种或多种第一蛋白质与一种或多种其它蛋白质之间的所述蛋白质相互作用。
溶解度可以通过用于研究蛋白质和蛋白质复合物的常规方法测定,所述常规方法包括用于捕获蛋白质和/或蛋白质结合配偶体、检测蛋白质和/或蛋白质结合配偶体、鉴定蛋白质和/或蛋白质结合配偶体、测试热稳定性或其任意组合的方法。例如,捕获蛋白质和/或蛋白质结合配偶体的方法包括,但不限于,亲和纯化、免疫沉淀、共免疫沉淀、染色质免疫沉淀,核糖核蛋白免疫沉淀、或其任意组合,其已经用于沉淀蛋白质和蛋白质复合物。检测蛋白质和/或蛋白质结合配偶体的方法可包括但不限于免疫检测测定、荧光测定、免疫染色、比色蛋白质测定或其任意组合。蛋白质和/或蛋白质结合配偶体的鉴定可通过质谱(ms)及其变型如串联ms、热电离ms、maldi-tof或加速器ms进行。测定蛋白质热稳定性的方法是常规的并且是本领域已知的,其可以包括但不限于热位移测定(thermalshiftassay)、dsf-gtp技术、thermofluor测定、内在色氨酸荧光寿命测定、内在色氨酸荧光波长测定、静态光散射测定、快速平行蛋白水解(fastpp)测定、尺寸排阻色谱(sec-ts)、荧光检测尺寸排阻色谱(fsec-ts)、放射性配体结合热稳定性测定(radioligandbindingthermostabilityassay)或其组合。最近,已经发现细胞热位移测定(cetsa)作为一种定量配体诱导的蛋白质热稳定的方法。甚至更近地,在称为质谱-细胞热位移测定(ms-cetsa),也称为热蛋白质组分析(tpp),的新方法中,ms用于与cetsa进行多重定量。ms-cetsa允许通过定量蛋白质的热稳定性对蛋白质-配体结合进行蛋白质组水平的评估,其中ms-cetsa数据通常以熔解曲线的形式呈现。在一个实例中,溶解度可通过但不限于ms-cetsa、ms、cetsa、免疫沉淀、共免疫沉淀或其任意组合来测定。在一个优选的实施方案中,溶解度通过质谱法获得。在另一个实例中,可以使用不含外源配体的对照样品通过ms-cetsa测定溶解度。在一个实例中,通过计算方法确定溶解度,其中通过将蛋白质的ms读数除以参照样品的ms读数,计算每个样品中相对于参照样品的每种蛋白质的可溶性部分。计算值表示为
在一个实施方案中,可以将所述一种或多种样品中的一种或多种第一蛋白质的溶解度与一种或多种其它蛋白质的溶解度进行比较,并且可以确定所述一种或多种样品中的一种或多种第一蛋白质和一种或多种其它蛋白质之间的溶解度相似度以获得pca特征,其中溶解度相似度的非随机增加指示所述一种或多种样品中的所述一种或多种第一蛋白质与所述一种或多种其它蛋白质之间的蛋白质相互作用。在一个实例中,溶解度相似度的非随机增加具有p值<0.05。通常应当理解,p值越低,溶解度相似度的增加越不可能是随机的。
测定溶解度相似度包括将一种或多种第一蛋白质和一种或多种其它蛋白质的溶解度与参照进行比较。用于测定溶解度相似度的参照可以是一组蛋白质的溶解度相似度。在一个实施方案中,参照是相同样品或不同样品中随机蛋白质组的溶解度相似度。在另一个实施方案中,参照是不同样品中的一种或多种第一蛋白质和一种或多种其它蛋白质的溶解度相似度。在一个实例中,参照是来自corum数据库的蛋白质复合物。在另一个实施方案中,参照可以是预测的或推测的蛋白质复合物。
pca特征是通过计算方法获得的,其中包括诸如但不限于温度、解折叠事件、共聚集事件、溶解度曲线、熔解曲线、溶解度、时间或其任意组合的参数的数据被用于导出pca特征。这可以包括使用不同的算法,其中算法可以包括机器学习分类器或者在不训练数据集的情况下被评分。在一个实例中,通过对已知的蛋白质-蛋白质相互作用数据评分并使用现有的网络聚类算法来鉴定已知的和推定的蛋白质复合物,获得pca特征。在另一个实例中,通过用pca导出的分数对装配的蛋白质-蛋白质相互作用网络中的每个相互作用的蛋白质对进行加权,并评估它们辅助网络/图形聚类算法(例如但不限于cmc、clusterone、mcl或其任意组合)鉴定corum中注释的蛋白质复合物的能力,获得pca特征。在又一实例中,通过使用机器学习分类器来集成多个正交数据从而获得pca特征,并利用诸如corum和string的金标准来校准以获得最优结果。在另一个实例中,通过仅使用pca对每个相互作用的蛋白质对进行评分而获得pca特征,而不使用训练数据集或金标准进行校准。在又一实例中,通过使用其它网络/图形聚类算法(例如但不限于coach、ipca或接受未加权网络作为输入的任何其它算法)来获得pca特征,随后根据它们的加权密度对所预测的复合物进行排序。在另一个实例中,通过对coach预测的蛋白质复合物进行基于pca的评分获得pca特征。
在一个实施方案中,可以使用一种或多种第一蛋白质和一种或多种其它蛋白质的溶解度之间的平均距离来确定该一种或多种第一蛋白质和一种或多种其它蛋白质之间的溶解度相似度。应当理解,平均距离可以在一个或多个温度或数据点测量。可以使用一种或多种距离度量来测定平均距离,并使用所述距离作为推定的相互作用蛋白质和非相互作用蛋白质之间的溶解度相似度的反测度(inversemeasure)。在一个实例中,距离度量可以包括但不限于欧式距离(euclideandistance)、皮尔逊相关系数(pearson’scorrelationcoefficient)、余弦相似度、曼哈顿距离(manhattandistance)、闵氏距离(minkowskidistance)、jaccard相似度、马氏距离(mahalanobisdistance)或切比雪夫距离(chebyshevdistance)。在一个优选实施方案中,距离度量是欧式距离或皮尔逊相关系数。在一个实例中,然后用如欧式距离或皮尔逊相关系数的度量来计算蛋白质对在温度d和时间t的每个或子集中的溶解度相似度。蛋白质x和y之间在温度d和时间t的每个或子集中的溶解度相似度表示为e(ts)x,y。计算值与相互作用概率和相互作用化学计量(蛋白质彼此相互作用的部分)相关。推导已知相互作用的蛋白质对和非相互作用蛋白质对之间溶解度相似度e(ts)的分布,以基于它们的溶解度相似度计算两种蛋白相互作用的概率。在另一个实例中,对于每种已知或预测的蛋白质复合物,包括二元蛋白质-蛋白质相互作用,复合物的每个亚基对的e(ts)如所述进行测定。对于具有多于两个亚基的蛋白质复合物,在亚基对之间计算的溶解度相似度的差异允许区分具有可能更高或更低相互作用/丰度化学计量的亚基对或亚复合物。蛋白质对x1x2和蛋白质对y1和y2的溶解度相似度的差异(表示为f)计算为|e(ts)x1,y1-e(ts)x2,y2|。通过推导随机选择的两个蛋白质对之间溶解度相似度差异的分布来评估以v计算的f的统计学显著性。经验p值计算为随机选择的蛋白质对与实际亚基对相比具有相等或更高f值的频率。可以计算每个复合物的所有亚基对中e(ts)的平均值和标准偏差,分别表示为a和sd。在另一个实例中,为了鉴定在不同生物样品/条件(已经以不同的温度被加热不同的持续时间)中差异调节的蛋白质复合物,推导出针对两个生物样品或条件之间的相同复合物计算的a和sd的差值。对于生物样品/条件i和j,差异表示为d(a)i,j和d(sd)i,j。d(a)i,j和d(sd)i,j的统计学显著性通过随机采样n种蛋白质来估算,其中n是在两种生物样品/条件下具有溶解度数据的蛋白质复合物的亚基数量。计算随机选择的蛋白质的d(a)i,j和d(sd)i,j。经验p值计算为随机选择的蛋白质集合具有等于或高于实际蛋白质复合物的d(a)i,j和d(sd)i,j的频率。
在第二方面,本发明涉及根据本发明的方法用于筛选治疗药物靶标、疾病进展预后、测定药物治疗的功效、测定蛋白质化学计量、鉴定新的蛋白质途径、绘制蛋白质相互作用组图谱、预测蛋白质功能或绘制时阈蛋白质相互作用图谱的用途。本文公开的方法可以以各种方式使用。在一个实例中,该方法可用于(但不限于)生物学、化学、物理学、分子生物学、细胞生物学、蛋白质组学、免疫学、药理学、结构生物学、生物物理学或生物化学领域。在另一实例中,该方法可用于但不限于药物筛选、筛选治疗性药物靶标、疾病进展预后、测定药物治疗的功效、测定蛋白质化学计量、鉴定新的蛋白质途径,绘制蛋白质相互作用组图谱、预测蛋白质功能、绘制时阈蛋白质相互作用图谱、对未保存在裂解物中的弱或瞬时蛋白质-蛋白质相互作用进行细胞内研究、验证蛋白质复合物或直接在非工程化细胞中研究蛋白质复合物和相互作用的化学调节剂。
本文说明性描述的本发明可以在不存在本文未具体公开的任何一个或多个要素、一个或多个限制的情况下适当地实施。因此,例如,术语“包含(comprising)”、“包括(including)”、“含有(containing)”等应被广泛地而非限制性地理解。另外,本文所采用的术语和表达已被用作描述术语而非限制术语,且在使用这些术语和表达时无意排除所展示和描述的特征或其部分的任何等效物,但应认识到,在所主张的本发明的范围内可进行各种修改。因此,应当理解,尽管已经通过优选实施方案和可选特征具体公开了本发明,但是本领域技术人员可以对本文公开的其中实施的发明进行修改和改变,并且这些修改和改变被认为在本发明的范围内。
如在本申请中使用的,单数形式“一种(a、an)”和“该(the)”包括复数引用,除非上下文另外明确地指出。例如,术语“遗传标记(geneticmarker)”包括多个遗传标记,包括其混合物和组合。
如本文所用,在制剂组分浓度的上下文中,术语“约(about)”通常是指所述值的+/-5%、更通常为所述值的+/-4%、更通常为所述值的+/-3%、更通常为所述值的+/-2%、甚至更通常为所述值的+/-1%、甚至更通常为所述值的+/-0.5%。
遍及本公开,某些实施方案可以范围形式公开。应当理解,范围形式的描述仅仅是为了方便和简洁,而不应被解释为对所公开范围的范畴的非灵活限制。因此,应当认为范围的描述已经具体公开了所有可能的子范围以及该范围内的各个数值。例如,对诸如1至6的范围的描述应当被认为已经具体公开了诸如1至3、1至4、1至5、2至4、2至6、3至6等的子范围,以及在该范围内的单个数字,例如1、2、3、4、5和6。无论范围的宽度如何,这都适用。
某些实施方案也可以在本文中广泛地和一般性地描述。落入一般公开内的较窄种类和亚类分组中的每一个也形成本公开的一部分。这包括具有附带条件或否定限制(从该类中去除任何主题,而不管所去除的材料是否在本文中具体列举)的实施方案的一般性描述。
本发明在此已被广泛地和一般性地描述。落入一般公开内的较窄种类和亚类分组中的每一个也形成本发明的一部分。这包括具有附带条件或否定限制(从该类中去除任何主题,而不管所去除的材料是否在本文中具体列举)的本发明的一般性描述。
其它实施方案在以下权利要求和非限制性实施例中。此外,在根据马库什群组描述本发明的特征或方面的情况下,本领域技术人员将认识到,由此也根据马库什群组的任意单个成员或成员子群组来描述本发明。
实验部分
通过参考具体实施例,将进一步更详细地描述本发明的非限制性实施例和比较例,这些实施例不应被解释为以任何方式限制本发明的范围。
材料和方法
cdk2和细胞周期蛋白e1实验
细胞转染
通过磷酸钙方法,在8小时期间,用总共12μg的质粒转染2x106hek293ft细胞,所述质粒编码带有人v5标签的细胞周期蛋白e1(pbobi-hs细胞周期蛋白e1--v5-pkb057)和/或带有ha标签的cdk2(pcmv-hscdk2-ha-pkb407)和/或mcherry(pbobi-mcherry-pkb1847)。48小时后通过在pbs中刮取细胞来收获细胞,在这之后对细胞进行计数并等分用于完整细胞cetsa实验。
蛋白质印迹和免疫检测
将来自转染293t细胞的10μg或25μg蛋白质提取物在9%聚丙烯酰胺凝胶上分离,使用半干系统将其转移到聚偏二氟乙烯膜(pvdf,millipore,#ipvh0010)上并在含有0.1%tween20和4%脱脂奶粉(bio-rad,#1706404)的tris缓冲盐水(tbs)中封闭。在4℃下用适当的一抗探测印迹过夜(抗v5,invitrogen#r960-25;抗ha,使用ha表位作为抗原制备针对ha的兔多克隆抗血清),随后使用缀合辣根过氧化物酶的二抗山羊抗小鼠(pierce,#0031432)或抗兔抗体(pierce,#0031462),并使用增强的化学发光(perkinelmer,#nel105001ea)显影。
免疫沉淀以少量修改进行。简言之,将50μg来自转染的293t细胞的蛋白质提取物与蛋白a琼脂糖珠(invitrogen#15918-014)在ebn缓冲液(80mmβ-甘油磷酸盐ph7.3,20mmegta,15mmmgcl2,150mmnacl,0.5%np-40,1mmdtt和蛋白酶抑制剂[亮抑肽酶、胰凝乳蛋白酶抑制剂和胃酶抑制剂(chemicon,ei8,ei6和ei10)各20μg/ml)])中在4℃孵育5小时,所述蛋白a琼脂糖珠与针对v5标签(invitrogen#r960-25)或ha标签(如上所述)的抗体预偶联。孵育后,在ebn缓冲液中洗涤珠子三次,将其干燥并重悬于1x浓缩的laemmli缓冲液中,然后准备好上清液在聚丙烯酰胺凝胶上进行电泳分离。
cetsa与脱盐实验
对于完整细胞cetsa实验,收获细胞,洗涤34并重悬于pbs中。处死小鼠后,立即通过门静脉用pbs灌注c57bl/6小鼠的肝脏。收集灌注的肝脏并使组织通过具有crpmi的65μ细胞过滤器以获得单细胞悬浮液。然后将单细胞悬浮液用pbs洗涤一次,然后再悬浮在pbs中以获得4000万细胞/ml的细胞浓度。然后将来自细胞系或小鼠肝脏的细胞分成10等份,在96孔热循环仪中以3℃间隔从37℃加热至64℃,持续3分钟,然后在4℃进行3分钟。通过加入2x激酶缓冲液(100mmhepes,ph7.5、10mmβ-甘油磷酸盐、0.2mm原钒酸钠(na3vo4)、20mmmgcl2、4mmtcep、2x蛋白酶抑制剂混合物)在热处理后裂解细胞,随后用液氮进行两次冻融循环并用注射器进行机械剪切,然后进行最后的冻融循环。通过在4℃以20,000g离心20min除去细胞碎片和聚集的蛋白质团粒。然后将上清液用于质谱样品制备。对于裂解物cetsa实验,首先用1x激酶裂解缓冲液(含有50mmhepes,4℃ph7.5、5mmβ-甘油磷酸盐、0.1mm活化原钒酸钠、10mm氯化镁、2mmtcep和无edta蛋白酶抑制剂),由4000万k562细胞生成1mlk562裂解物。用2x冻融完全裂解细胞,然后用21”规格针进行10x机械剪切,用30”规格针进行3x机械剪切,最后冻融。裂解物以20,000g在4℃旋转减慢20min。保留上清液,等分后冷冻并储存在-80℃。在cetsa加热实验之前,将等分试样在室温下完全解冻并达到至室温。
k562裂解物脱盐
用1x激酶裂解缓冲液平衡pd-10脱盐柱(gehealthcare,产品编号:17-0851-01)。随后,将1mlk562裂解物加入pd-10柱中,随后用1.5ml1x激酶裂解缓冲液补足。接着,用0.5ml1x激酶裂解缓冲液进行洗脱步骤,达7次。使用蛋白浓度最高的7个级分中的2个级分,将其合并得到1ml脱盐的裂解物,用于cetsa实验。
甲氨蝶呤与s-阶段实验
k562以0.25x106细胞/ml铺板并用媒介物(dmso)或溶在dmso中的20μm甲氨蝶呤(sigma)处理48小时。收集细胞,将其洗涤、重悬于冰冷的pbs缓冲液中,随后等分成10个级分。遵循标准完整细胞cetsa和裂解方案。
nfκb和甲氨蝶呤实验
蛋白质印迹
甲氨蝶呤(20μm)处理48小时和/或tnf-α(10ng/ml)处理30分钟后收获细胞。细胞用dpbs(无钙和镁)洗涤一次,然后在totex缓冲液(20mmhepes,ph7.9、0.35mnacl、20%甘油、1%np-40、1mmmgcl2、0.1mmegta)中裂解,所述缓冲液包括50mmnaf、0.3mmna2vo3和蛋白酶抑制剂混合物。收集包括蛋白质的上清液,然后以全速离心20min。使用以下抗体通过以下标准程序进行蛋白质印迹:兔抗-磷酸-ikkα/β(s180/181)(1:1,000;bioworldtechnology,bs5082)、兔抗-磷酸-ikbα(s32)(1:1,000;cellsignallingtechnology,2859)、兔抗-ikbα(1:1,000;santacruzbiotechnology,sc-371)、兔抗-磷酸-p65(s536)(1:1,000;cellsignallingtechnology,3031)、鼠抗-hsp90(1:1,000;bd,610419)、山羊抗兔igg-hrp(1:5,000;santacruzbiotechnology,sc-2054)和山羊抗鼠igg-hrp(1:5,000;santacruzbiotechnology,sc-2005)。
共免疫沉淀
收获的细胞在包括蛋白酶抑制剂混合物的免疫沉淀裂解缓冲液(ph8.0的50mmtris、1%tritonx-100、0.5%np-40、170mmnacl、5%甘油、1%edta)中裂解并超声处理5分钟(打开30秒,关闭30秒)。离心后收集包含蛋白质的上清液并测量蛋白质浓度。使用每个样品的1mg蛋白进行免疫沉淀。将样品与pureprotome蛋白a磁珠(millipore,lskmaga10)孵育1小时至预澄清(pre-clear),然后去除磁珠,将样品与下述抗体孵育过夜:兔抗nf-κbp65(5μg/mg样品;santacruz,sc-372)、兔抗nf-κbp50(5μg/mg样品;santacruz,sc-7178)和正常兔igg(5μg/mg样品;santacruz,sc-2027)。孵育后,加入磁珠以捕获抗体-蛋白质复合物,持续1小时,然后使用sds上样缓冲液从磁珠上洗脱蛋白质。使用以下抗体将样品用于蛋白质印迹:兔抗-ikbα(1:1,000;santacruzbiotechnology,sc-371)、兔抗nf-κbp65(1:1,000;santacruz,sc-372)、兔抗nf-κbp50(1:1,000;santacruz,sc-7178)和verilblot二抗(hrp)(1:10,000;abcam,ab131366)。
细胞培养
k562和a375在rpmi培养基(gibco,产品编号:a10491)中培养,hek293t和mcf7在dmem培养基(gibco,产品编号:11995)中培养,hl60在imdm培养基(gibco,产品编号:12440)中培养,且hct116在mccoy培养基(gibco,产品编号:12330)中培养。除补充有20%胎牛血清的imdm培养基外,所有培养基都补充了10%胎牛血清(biowest,产品编号:s1810)。rpmi和dmem进一步补充有l-谷氨酰胺(gibco,产品编号:25030)、非必需氨基酸(gibco,产品编号:11140)和青霉素链霉素(gibco,产品编号:15140)。所有贴壁细胞系均用胰蛋白酶edta(0.25%)酚红(gibco,产品编号:25200)传代或收获。
ms样品制备及数据处理
在20,000g,4℃离心10min或20min后收集上清液并用bca测定定量可溶性蛋白质。蛋白质在0.1%(w/v)rapigesttmsf表面活性剂(waters)溶液中变性,在55℃下用20mmtcep还原20min,然后在室温下用55mm氯乙酰胺烷基化30min。将蛋白质用lys-c(wako)在37℃预消化3-4小时,然后用胰蛋白酶(promega)在37℃消化过夜。用1%tfa将样品降至ph2以水解rapigesttm并通过离心进行团粒化。从上清液收集消化的样品并在真空浓缩器(eppendorf)中完全干燥。然后将样品溶解在100mm或200mmteab缓冲液(ph8.5)中,并根据制造商手册用tmt10plex等压标记试剂(pierce)标记。标记反应在室温下进行1-2小时,然后用1mtris缓冲液(ph7.4)猝灭。根据标记集(labelset)并标记的样品,将其酸化并在c18sep-pak盒(waters)上脱盐。将脱盐的样品干燥,再悬浮在5%氢氧化铵溶液(ph10.0)中,然后使用高ph反相zorbax300extend-c18柱(5μm,4.6mm×250mm,134agilent)在
与qexactivetm或qexactivetmhf质谱仪(thermofisherscientific)偶联的dionexultimate3000rslcnano系统(thermofisherscientific)上,在50cm×75umeasy-spraytmc18lc柱(thermoscientific)上,以80min梯度的溶剂a(0.5%乙酸水溶液)和溶剂b(80%acn,0.5%乙酸水溶液)分离每个注射样品。以数据依赖性采集采集数据。每个ms扫描中的前12个峰均经受更高的能量碰撞解离(hcd)断裂。ms1扫描分辨率为70,000(m/z=200th),而ms/ms扫描分辨率为35,000。
使用thermoproteomediscoverer软件(2.0.0版,thermofisherscientific)产生峰列表。使用mascot和setquest针对目标诱饵人hhv4uniprot数据库进行光谱搜索。将脲甲基半胱氨酸和标记于肽n-末端上的tmt10plex以及赖氨酸设定为固定修饰。将氧化(m)、脱酰胺基(nq)和乙酰化蛋白质n-末端设置为可变修饰。对于肽分配,需要最小6个氨基酸的长度和最多3个错切(missedcleavage),同时分别允许ms全谱扫描的最大30ppm质量偏差和ms/ms离子片段的0.06da质量偏差。在psm和肽水平上对于高置信度肽以0.01的水平且对于中等置信度肽以0.05的水平进行fdr对照。报告离子定量的共隔离(co-isolation)阈值设定为50%。装配蛋白质群组用于下游数据分析。
ms数据后处理与生物信息学分析
数据归一化与曲线拟合
将来自每个样品的每个蛋白质的报告离子强度除以来自37℃样品(裂解物或完整细胞)的相同蛋白质的报告离子强度,以获得蛋白质在不同温度下的标准化溶解度。这些来自每个ms运行的每种蛋白质的读数均源自其根据比率计数(用于定量的psm数目)加权的同种型(例如pxxxxx-1,pxxxxx-2)的平均读数。计算出的平均值的比率计数被计算为比率计数之和。从每个技术和生物学重复的ms运行中鉴定出的蛋白质中除去常见污染物和片段序列的uniprot登录号。接下来,对于每个温度,计算蛋白质在重复中的溶解度读数的中位数(mom)。然后,使用十个温度的mom导出s形曲线,该曲线使用drc(剂量-响应曲线分析)r包与三参数对数逻辑函数(ll3.u,其中上限为1.0)拟合。该s形曲线可以解释为人蛋白质组在不同温度下的一般溶解行为。假定该一般溶解度在重复中是保守的,因此每个重复的每个温度的标度读数相应地显示出这一点。具体地,每种蛋白质的溶解度读数都通过每个样品和温度的固定值进行调整,以使每个温度下的中值读数现在落在导出的s形曲线上。为了合并用于分析的重复,将重复中相同蛋白质的溶解度读数平均化,通过相应求和的比率计数进行加权。每种蛋白质在10个温度下的溶解度值分别与r拟合为四次函数,以得出熔解曲线,从而进行可视化和检查。
pca特征的经验统计评估
为了鉴定显示pca行为的已知人蛋白质复合物,对于每种蛋白质复合物,计算蛋白质复合物的所有亚基对之间的熔解曲线之间的平均欧式距离(表示为eavg)。随机采样具有相同蛋白质数量的10,000个随机蛋白质集合,并计算每个随机集合的eavg。然后将观察到的蛋白质复合物的eavg的经验p值确定为随机蛋白质集合产生等于或小于蛋白质复合物的eavg的eavg的频率。具体地,令pa=(p1,p2…,pn)为具有溶解度数据的蛋白质复合物a的n个唯一亚基的集合。来自pa的所有唯一亚基对之间的平均欧式距离(eavg)被计算为
其中m(来自pa的唯一亚基对的数量)等于(n2–n)/2,且d(px,py),蛋白质px和py的熔解曲线之间的欧式距离计算为
其中(x1、x2、x3、x4、x5、x6、x7、x8、x9、x10)和(y1、y2、y3、y4、y5、y6、y7、y8、y9、y10)分别表示蛋白质px和py在37℃、40℃、43℃、46℃、49℃、52℃、55℃、58℃、61℃和64℃下的标准化溶解度。为了估算获得与eavg(pa)相同或更小的值的概率,计算10,000个随机选择的大小为n的蛋白质集合的eavg。观察到的eavg(pa)的经验p值被确定为eavg≤eavg(pa)的随机蛋白质集合的频率。
观察到的pca特征变化的经验统计评价
如果pa=(p1,p2…,pn)是来自具有来自dmso处理和甲氨蝶呤处理的k562样品的溶解度数据的复合物a的整个亚基集合,则为pca特征的差异计算如下
δ(dmso-mtx)=eavg(pa(dmso))-eavg(pa(mtx))(3)
其中eavg(pa(dmso))和eavg(pa(mtx))由分别来源于dmso处理的样品和甲氨蝶呤处理的样品的溶解度数据计算。为了评价观察到的δ(dmso-mtx)的统计学显著性,计算了大小为n的10,000个随机选择的蛋白质集的δ。然后将观察到的δ(dmso-mtx)的统计学显著性估算为导致δ≤δ(dmso-mtx)的随机蛋白质集合的频率。
用pca导出的z分数加权的相互作用网络的构建
构建对每个细胞系特异性的加权相互作用网络。每个报道的蛋白质-蛋白质相互作用均用细胞特异性ms-cetsa数据加权。首先,为每个报道的蛋白质-蛋白质相互作用计算欧式距离的导数ex=1/(1+e),其中e是两个相互作用的蛋白质的熔解曲线之间的欧式距离(参见等式2)。ex将以更对称的分布限制在0和1之间。接下来,将每种蛋白质相互作用的z-分数导出为(ex-μ)/σ,其中μ和σ是从在每种细胞系中鉴定的全部蛋白质对计算的ex的平均值和标准偏差。该z-分数允许在各细胞系间直接比较pca特征。为了便于鉴定每种细胞系中可能的差异蛋白质-蛋白质相互作用,对于每种蛋白质-蛋白质相互作用,通过去除最高和最低z-分数后的六个细胞系的z-分数的平均化来构建基础相互作用网络。
蛋白质之间相互作用化学计量及丰度化学计量的分析
将相互作用化学计量和丰度化学计量均相对于诱饵蛋白标准化。为了分析的目的,替代地,将化学计量标准化为更高丰度的蛋白质。具体地,如果相互作用蛋白的相互作用化学计量f或丰度化学计量a大于1,则将其分别转化为1/f或1/a。
差异蛋白表达分析
limmar包用于鉴定来自两个生物学重复集的dmso处理的和甲氨蝶呤处理的k562细胞之间差异表达的蛋白质。来自原始cetsa/tpca实验的37℃处理的上清液样品被加工,用于如前所述利用tmt等压标记试剂(pierce)进行ms分析。原始ms数据如前所述进行处理。对样品x特异性的比例因子(表示为sfx)计算为ix/min{i1,i2,i3,i4},其中x∈{1,2,3,4}并且ix是样品x的全部蛋白质的总报告离子强度。随后,样品x中每种蛋白质的报告离子强度除以sfx。然后将数据对数转换并作为单通道数据类型输入到limma包中,将甲氨蝶呤处理的样品与dmso处理的样品进行对比,并通过“fdr”调节p值以进行多重假设检验。
实施例1
相互作用的蛋白质对表现出强pca特征
用充分表征的cdk2-细胞周期蛋白e1复合物研究pca,使用免疫印迹导出在人胚肾(hek)293t细胞中过表达的两种蛋白质的熔解曲线(图24a和图24b)。单独表达的细胞周期蛋白e1-v5和cdk2-ha(血凝素)显示不同的熔解曲线,但是当共表达时,它们相互作用并以相似的熔解曲线稳定(图2和图24a),这与pca假说一致。
为了评估pca的通用性,从相同k562裂解物的8个ms-cetsa实验中核对7693种人蛋白质的熔解曲线(图3)。在7693种蛋白质之间,111,776种蛋白质-蛋白质相互作用被装配并在biogrid、inact和mint数据库中注释。使用欧氏距离作为曲线相似度的反测度,观察到相互作用的蛋白质对通常比全部蛋白质对具有更高的曲线相似度(p<2.2×10-16,单尾mann-whitney检验,图4a)。这种相似度对于多个出版物报道的相互作用更显著(图4a和图25)。对于最近两项使用酵母双杂交(y2h)和亲和纯化(ap)-ms/ms进行的大规模研究中的相互作用,以及对于corum数据库中的复合物的亚基对,也观察到了高的曲线相似度(图4b)。因此,观察到的pca特征不太可能由所使用的确认偏倚(ascertainmentbias)和实验方法引起。
计算每种蛋白质复合物的所有亚基对之间的平均曲线相似度,并在蛋白质复合物水平评估pca特征。对于含有至少三个具有熔解曲线的亚基的558种非冗余人复合物,在所有亚基中总共有160种表现出非随机pca特征(p<0.05,图5)。
实施例2
pca特征在来自完整细胞的数据中较强
接着,从通过脱盐耗尽低分子量(lmw)配体的k562细胞裂解物中获得ms-cetsa数据。对于大多数蛋白质复合物(约88%,图26a),观察到降低的平均曲线相似度,表明lmw配体耗尽引起复合物解离增加。然而,观察到pca变化可忽略不计但所有亚基的熔解曲线相似偏移的多种蛋白质复合物,例如pa700蛋白酶体亚复合物,其可能是由于腺苷5′-三磷酸(atp)耗尽所致(图6和图27)。因此,全蛋白质复合物可保持完整,但由于lmw配体耗尽而热不稳定。相比之下,来自新批次k562裂解物的ms-cetsa数据与先前裂解物均具有良好的再现性(皮尔逊相关系数(pearson’sr)=0.88)。对于在每个数据集中具有至少三种定量肽的蛋白质,观察到更高的再现性(图26c),因此组合了现有的和新的数据集,用于随后的分析。
对完整k562细胞进行6次ms-cetsa实验。观察到在裂解物数据中全部蛋白质对的熔解曲线在统计学上更相似(图7),而相互作用蛋白质对的熔解曲线在完整细胞数据中在统计学上更相似(图7)。还观察到蛋白质复合物在完整细胞数据中比在裂解物数据中具有更好的亚基间的平均曲线相似度(图28a-d)。总之,完整细胞数据显示出比裂解物数据更多的具有非随机pca特征的蛋白质复合物(图8、图29和图30)。
相比裂解物数据,完整细胞数据在统计学上具有丰富的(在应激反应、细胞周期和dna加工途径方面)pca显著的蛋白质复合物(图31)。许多dna-染色质相关复合物和膜相关复合物——分别以nup107-160核孔亚复合物和起始识别复合物(图9)为例——仅在完整细胞中表现出显著的pca特征,推测是由于细胞裂解物中dna-染色质和天然膜的破坏。分析表明,复制因子复合物c(rfc)在完整细胞和裂解物中都保持装配(图10a,上图),但整个复合物在裂解物中热不稳定(图10a,左上图),推测可能是由于没有dna-染色质。
观察到许多情况,其中亚复合物在完整细胞和裂解物数据集之间显示不同的pca特征。例如,40s和60s核糖体亚复合物各自含有具有类似曲线的亚基,其在完整细胞数据中更靠近其它亚复合物移位(图10b),推测可能是因为更完全装配的核糖体在细胞中而不是在裂解物中翻译mrna。另一个实例是具有基于熔解曲线的两个不同亚基群组的ndc80着丝粒复合物,所述熔解曲线在完整细胞数据中比在裂解物数据中彼此更相似(图10a,底部)。两个不同的亚基群组对应于通过短四聚蛋白区彼此缔合的两个稳定的异二聚体(图11)。pca分析表明两个亚复合物在裂解物中解离。这些观察表明pca可以揭示蛋白质复合物结构及其功能状态的基本差异。
实施例3
相互作用的蛋白质之间的pca特征相互作用化学计量和丰度化学计量。
复合物中亚基之间的相互作用化学计量和丰度化学计量可潜在影响其pca特征。针对报告具有最高置信度的相互作用的蛋白质对分析这些数据,并观察到pca特征与相互作用化学计量(斯皮尔曼相关系数(spearman’sr)=-0.22,p<0.001,图12a和图32a)和丰度化学计量(斯皮尔曼相关系数=-0.21,p<0.001,图12b和图32b)正相关。相互作用化学计量和丰度化学计量均大于80%的蛋白质对表现出比分别满足两个标准的蛋白质对更强的pca特征(图12c)。相比之下,仅通过相似丰度配对的蛋白质不显示强pca特征(图33a至c)。因此,将相互作用化学计量与丰度化学计量相乘产生与pca特征更好相关的值(斯皮尔曼相关系数=-0.27,p<0.001,图12d和图32c)。因此,相互作用的蛋白质之间的相互作用化学计量和丰度化学计量被pca固有地捕获,尽管是半定量的。
在核心-附着模型下,蛋白质的子集形成蛋白质复合物的稳定核心,所述蛋白质复合物被其它蛋白质差异地或暂时地结合。用pca研究该模型,比较来自复合物核心的蛋白质对的熔解曲线与核心和附着亚基之间的蛋白质对的曲线(图13)。核心-核心对在每个复合物中鉴定为在两个或多个其它corum复合物中发现的那些,且核心-附着对鉴定为仅在该复合物中发现的那些。对于大多数复合物,核心-核心对的pca特征与核心-附着对的pca特征相似或比其强得多(图14)。这与核心-附着模型一致,在该模型中核心-附着相互作用通常在较低化学计量下发生。因此,pca特征可能概括了许多复合物的暂时附着和核心-附着结构。
接着,探索pca用于全系统监测细胞状态之间的蛋白质复合物动力学。完整的细胞ms-cetsa数据是从利用甲氨蝶呤在细胞周期s期停滞的k562细胞(图34)和从二甲基亚砜(dmso)处理的未同步细胞中获得的。对任何corum复合物观察到的pca增强(即更高的平均曲线相似度)进行了统计学显著性评估(参见材料和方法)。鉴定出18种蛋白质复合物(其中一些包含重叠的亚基)在s期同步化细胞的两个生物学重复中具有统计学增强的pca特征。
除3种鉴定的受调节蛋白质复合物外,所有这些以前都涉及s期。它们包括:在s期形成并定位到复制叉的caf-1复合体(图15);复制叉进程所需的trex-tho复合物(图15);以及s期进程所需的trap复合物。还鉴定了许多染色质-核小体重塑和相关复合物,如baf(图15)、larc和brg1-sin3a,与s期染色质的预期重塑一致。组蛋白mrna在s期之前上调,但随着dna复制停止而迅速降解。三种受调节的蛋白质复合物——mrna衰变复合物、外来体和整合子(integrator)复合物——被鉴定为参与组蛋白mrna降解。据报道,外来体的亚基rrp6的缺失在s期期间增加组蛋白mrnahtb1,因此突出了该复合物在s期期间的作用。整合子复合物涉及复制依赖性组蛋白mrna的加工。
不清楚是否涉及s期的三种鉴定的复合物中的两种是mdc1-mrn-atm-fancd2和dna连接酶iii-xrcc-pnk-聚合酶iii复合物。然而,根据已知的甲氨蝶呤的dna损伤作用,它们参与dna损伤应答。不涉及s期的第三种复合物是肿瘤坏死因子-α(tnf-α)-核因子kβ(nf-kβ)信号转导复合物(图15)。在tnf存在和不存在的情况下,证明通过抑制k562细胞中的iκb激酶α/β(ikkα/β)复合物、iκbα和p65的磷酸化,氨甲喋呤通过抑制nf-kβ信号转导途径的活化来增加相关的ikβα-p65-p50蛋白质复合物的装配(图16a和b)。来自两个重复的数据随后被合并——更好的数据覆盖和精度允许在s相和非同步化细胞之间更多被差异调节的复合体被发现。
实施例4
多个细胞系中的pca特征揭示细胞特异性相互作用,并且预测蛋白质复合物。
所鉴定复合物的增强pca特征主要源于大多数亚基的曲线收敛(图17)。然而,这可能由蛋白质表达的同步变化引起。因此,使用ms定量dmso处理的和甲氨蝶呤处理的细胞之间的相对蛋白质丰度。在生物学重复中观察到相对丰度的高再现性(皮尔逊相关系数=0.87,图35),并且仅鉴定了具有差异蛋白质表达的5个亚基(p<0.05,或p<0.1时的9个)(图17),所述5个亚基主要是介体(mediator)和整合子巨复合物(megacomplex)的部分。因此,大多数亚基在没有差异蛋白质表达的情况下彼此表现出增强的pca特征(图17)。
接着,在多个细胞系中分析pca特征。从hek293t、a375、hct116、mcf7和hl60完整细胞产生ms-cetsa数据。结合来自两个生物学重复的数据,所述两个生物学重复每个均进行两次技术性ms运行(生物学重复之间的平均皮尔逊相关系数=0.92),获得每个细胞系平均约7600种蛋白质的熔解曲线(图36a)。平均而言,33.8%的合格corum复合物在每个细胞系中表现出非随机pca特征(p<0.05)(图36b和图18),细胞系之间具有约70%的重叠。还获得了来自小鼠肝脏的数据,并观察到约37.7%的合格蛋白质复合物(三个或更多个具有熔解曲线的亚基)具有非随机pca特征,包括参与基础细胞过程的许多蛋白质复合物(图19)。因此,可在多种细胞系和组织样品中观察到pca。许多蛋白质复合物在6个细胞系中表现出强pca行为(图37),但也发现许多具有高质量但在各个细胞系中具有不同曲线的复合物(图38和39)。这表明,即使对于基本的和丰富的蛋白质复合物,细胞系之间蛋白质复合物化学计量和组成的合理(plausible)变化可能是由相互作用化学计量和/或蛋白质丰度的变化引起的。
许多蛋白质复合物在6个细胞系中表现出强pca行为,包括pa700蛋白酶体亚复合物和后期促进复合物。然而,发现了许多差异化的复合物,包括参与基础细胞过程的那些,如eif3核心复合物和前折叠素复合物。对照最近大规模相互作用研究的数据,观察到pca特征与蛋白质-蛋白质相互作用的化学计量相关(图12b)。因此,观察到的pca特征的差异表明,即使对于最基本和最丰富的复合物,蛋白质复合物的化学计量和组成也可以在细胞系之间变化。总计,观察到超过350个corum人蛋白质复合物在6个细胞系的至少一个中表现出显著的pca特征。
在所有6个细胞系中鉴定了报道的蛋白质间相互作用的加权网络,并产生了pca导出的z-分数。在去除每个相互作用的最高和最低z分数之后,还构建了平均化z分数的基础网络。将基础网络与细胞特异性pca加权网络进行比较有助于鉴定潜在差异化的相互作用、途径和功能模块。关注hct116,发现ras-raf-mek-erk途径含有许多具有高度差异化pca特征的相互作用(图20)。回顾性地,与其它细胞系相比,在hct116细胞中观察到braf和raf1蛋白的熔解曲线的显著相似度(图21),这与hct116中由活性krasg13d驱动的braf和raf1的预期二聚化一致。pca分析还表明braf-craf与mek的相互作用在hct116细胞中上调,但在表达brafv600e的a375细胞中下调(图20)。这与最近的发现(相互作用在表达突变体kras和野生型braf的细胞中上调,但在具有突变体braf的细胞中受抑制)相一致。因此,pca可能捕获细胞特异性相互作用和途径。
最后,使用图形或网络聚类算法和基于pca的相互作用评分,证实了已知的和可能先前未知的蛋白质复合物可以从现有的人相互作用组图谱(interactomemap)中鉴定(图22)。使用coach算法获得最佳性能,该算法结合了蛋白质复合物的核心-附着模型。使用该算法和基于pca的相互作用评分提供了与已发布的相互作用可靠性评分(包含出版物计数和功能相似度)相当的性能。还观察到,处于其当前形式的pca分析对蛋白质-蛋白质相互作用具有预测能力,其中根据相互作用数据集,曲线下面积的范围为0.62至0.79(图23a和b),这总体表明pca分析还可以与其它方法结合起来用于发现新的相互作用和蛋白质复合物。
实施例5
用pca评分和图形/网络聚类算法从蛋白质相互作用网络中鉴定推定的蛋白质复合物
研究pca特征是否可用于从装配的蛋白质-蛋白质相互作用网络预测已知的和验证的蛋白质复合物。具体地,用pca导出的分数(如1/(1+e),其中e是两个熔解曲线之间的欧式距离)对装配的蛋白质-蛋白质相互作用网络中的每个相互作用的蛋白质对进行加权,并且评估它们辅助三种网络/图形聚类算法(即cmc、clusterone和mcl)鉴定在corum中注释的蛋白质复合物的能力。这与许多以前的项目非常相似,这些项目对它们导出的蛋白质-蛋白质相互作用数据进行评分,并使用现有的图形/网络聚类算法来鉴定已知的和推定的蛋白质复合物。然而,与使用机器学习分类器整合多个正交数据并用金标准物如corum和string校准以获得最佳结果的先前项目不同,仅使用pca对每个相互作用的蛋白质对进行评分,而不使用训练数据集或用于校准的金标准物。还测试了接受未加权网络作为输入的两个其它网络/图形聚类算法coach和ipca,但是随后根据它们的加权密度(预测的复合物中的边线权重之和/预测的复合物中可能的边线的数量)对预测的复合物进行排序。
为了提高蛋白质复合物(特别是对于细胞或条件特异性的那些)的覆盖率,使用在基础状态下分析(profiled)的所有6个细胞系的pca数据,从来自分析的6个细胞系的两个最佳得分的平均值获得每个相互作用的最终pca导出分数。对于基线比较,每个相互作用的蛋白质对的可靠性/置信度基于其报道出版物的数量进行评分,所述报道出版物的数量被标准化为最高出版物计数。然而,使用由至少2个出版物报道的相互作用的蛋白质对,因为发现所形成的蛋白质网络有利于鉴定,并且在所用的5种网络/图形聚类算法中对corum复合物排序靠前。对于每个算法,测试相同的参数集,并为每个加权网络选择最佳auc(曲线下面积)。
观察到与基于出版物的评分相比,pca加权的蛋白质-蛋白质相互作用网络在所测试的5种算法中的3种中具有显著更好的性能,而对于其余的算法,性能是相当的。通过coach算法,获得对pca性能的最大程度的提高。有趣的是,coach算法通过先鉴定复合物核心,随后鉴定附着亚基,明确地利用了核心-附着模型来增强对蛋白质相互作用网络中的蛋白质复合物的鉴定。通过coach预测的复合物的基于pca的评分,丰富了已知和经过验证的corum复合物的最高命中(tophit)。通过coach鉴定出具有高覆盖率(鉴定出75%或更多亚基)并且通过pca排序靠前的复合物包括多合成酶复合物、pa700-20s-pa28复合物、mcm复合物、保守寡聚高尔基体(cog)复合物、浓缩蛋白复合物和trex/tho复合物。
接下来,将基于pca的评分与先前公开的相互作用可靠性评分方案进行比较,该方案组合了出版物计数和每种报道实验方法的一致性(coherency),以从相同的细胞组分和/或蛋白质复合物中鉴定相互作用的蛋白质对。不同实验方法的一致性计算为检测到的相互作用蛋白质对的分数,其中两种蛋白都具有高水平的细胞组分基因本体论(go)术语。对于每个相互作用的蛋白质对,使用noisy-or模型表示最终可靠性分数:
其中x(a,b)是检测蛋白质a和b之间的相互作用(a,b)的一组实验方法,而ne,(a,b)是实验方法e检测到相互作用(a,b)的次数。
观察到这5种算法的pca性能与使用相互作用可靠性评分方案非常相似,该方案结合了出版频率和间接的共定位或共复合物信息。因此,用现有的蛋白质-蛋白质相互作用数据进行pca评分可以在不同的网络/图形聚类算法中预测已知的和验证的蛋白质复合物,这与使用出版物计数和现有注释相当。此外,pca与其它正交证据的组合可以合理地进一步改善对注释蛋白质复合物的预测。
总之,pca使得能够在完整的非工程细胞和组织中同时进行多种蛋白质复合物的细胞内动力学研究。每个细胞系中约三分之一的合格corum复合物表现出非随机pca特征,其中在至少一个所分析的细胞系中58%的复合物表现出非随机pca特征。膜嵌入复合物包括在corum中,但可能不符合其它方案。具有非常低的相互作用化学计量的蛋白质复合物可能会产生不显著的pca特征——增加的装配可以通过pca特征的变化进行监测。pca分析表明,在不与dna和lmw配体如atp相互作用的情况下,许多复合物可以保持完整但热不稳定(图6和10)。观察到许多线粒体蛋白在完整细胞中更耐热。
该研究表明pca构成了一种用于蛋白质相互作用的全系统研究的方法。
等同物
前述实施例是为了说明本发明而给出的,不应解释为对本发明的范围施加任何限制。显而易见的是,在不脱离本发明基础原理的前提下,可以对本发明上面描述的并在实施例中说明的具体实施方案进行许多修改和改变。所有这些修改和改变都旨在被本申请所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!