化合物、试剂及其用途的制作方法
相关申请
本申请要求2017年9月28日提交的美国临时申请号62/564,558在美国法典第35篇第119(e)条下的提交日期的权益,其整个内容(包括所有附图、式、说明书和权利要求)通过引用并入本文。
背景技术:
提供以下描述发明背景的信息以辅助理解本发明,并且不被认为构成或描述了本发明的现有技术。
传统上使用肾小球滤过率(gfr)来评估肾功能。尽管黄金标准方法(测量的gfr(mgfr))是精确的和准确的,但由于试验的复杂、耗时和侵入性质,其实用性受到限制。它也是执行起来昂贵的试验。所以,在临床上使用基于在血清中测量的内源性过滤标志物肌酸酐和半胱氨酸蛋白酶抑制剂c的浓度的方程式来估计gfr。当前使用的用于估计gfr(egfr)(mdrd、ckd-epi)的方程式包括人口统计学变量(年龄、性别、种族)作为与肾功能/gfr不相关的因素的替代物,所述因素影响肌酸酐和半胱氨酸蛋白酶抑制剂c的血清水平。但是,这些egfr方程式显示出与mgfr相比有限的准确度和精确度。
当前的标志物可能具有有限的实用性,因为人口统计学变量对于非gfr决定因素(例如关于肌酸酐的肌肉质量、关于半胱氨酸蛋白酶抑制剂c的体脂肪率)而言是差的替代物,某些患者被不适当地分类(例如,多种族、变性、雌雄间体)以及对报告的当地敏感性(例如,种族)。由于当前egfr标志物的局限性,已经鉴定出由肾小球自由过滤且不受(或仅最小地受)非-gfr决定因素影响的具有低分子量的替代内源性过滤标志物。这些替代标志物也可以独立于人口统计学变量,从而克服与使用这些变量相关的限制。
本文描述了合成、分离、检测和定量生物样品中的内源性小分子过滤标志物((2r,3r)-2,3-二羟基-5-甲硫基-反式-4-戊烯酸,dmtpa)的方法。先前发现的分析物/标志物为x-11564,这是一种与gfr和肾功能相关的标志物,但结构未知,这限制了其实用性。在这里,我们报告了该分子的结构,并提供了该化合物的合成方法,测量方法(包括单独地或作为一组标志物的一部分测量),测量试剂(例如,内部标准品、抗体)以及使用它们的试剂盒。
技术实现要素:
本发明提供了新颖的化合物和组合物、合成方法以及用于检测生物样品中的化合物的量的方法,其可用于评估和监测肾功能以及作为关于肾脏疾病的诊断方法。
在一个实施方案中,本发明提供了由式(i)表示的化合物或其盐:
其中所述化合物是至少60%、70%、80%、90%、95%、99%、99.5%或99.9%纯的。
在另一个实施方案中,本发明提供了由式(ii)表示的化合物或其盐:
其中所述化合物是至少60%、70%、80%、90%、95%、99%、99.5%或99.9%纯的。
在另一个实施方案中,本发明提供了使用测定来确定式i或式ii的化合物或其盐的水平的方法。
在另一个实施方案中,本发明提供了用于确定受试者中式i或式ii的化合物或其盐的水平的方法,所述方法包括:(1)从得自所述受试者的生物样品制备分析样品;和(2)使用色谱法、质谱法、酶联免疫吸附测定(elisa)、抗体连接、免疫印迹法、免疫组织化学(ihc)、其它免疫化学方法或它们的组合确定所述化合物的水平。
在本发明的一个方面,一种方法包括通过质谱法检测和确定样品中式i或式ii的化合物或其盐的量。在另一个方面,一种方法包括通过质谱法检测和确定样品中式i或式ii的化合物或其盐和一组分析物中的一种或多种的量,所述一组分析物包含一种或多种选自假尿苷、n-乙酰基苏氨酸、苯基乙酰基谷氨酰胺、色氨酸、n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)、肌酸酐、中赤藓醇、阿糖醇、肌-肌醇、n-乙酰基丝氨酸、n-乙酰基丙氨酸、3-甲基组氨酸、反式-4-羟脯氨酸、犬尿氨酸、尿素、3-吲哚氧基硫酸盐和它们的组合的分析物。还提供了从生物样品提取分析物的方法和在检测之前通过质谱法色谱分离分析物的方法。
在本发明的一个方面,其中所述方法包括通过质谱法检测和确定样品中化合物a的量,化合物a的质谱图可以包含一个或多个选自约177、159、133、131、129、115、113、111、101、100、99、89、85、83、75、73、57.03、57.00、97、87、74、84和67的峰(即,质荷比(m/z))。
在本文中使用的术语“约”是指值±0.5。
在另一个实施方案中,本发明提供了用于计算受试者中的估计的肾小球滤过率(egfr)的方法,所述方法包括下述步骤:
1)使用色谱法、质谱法、酶联免疫吸附测定(elisa)、抗体连接、免疫印迹法、免疫组织化学(ihc)、其它免疫化学方法或它们的组合,确定在得自所述受试者的生物样品中由式i或式ii表示的化合物或其盐的水平;和
2)使用利用所确定的化合物水平的算法计算egfr。
在其它实施方案中,本发明提供了基于在得自所述受试者的生物样品中检测到的由式i或式ii表示的化合物的水平来确定受试者发生肾功能下降的倾向、根据肾功能的水平(或阶段)将受试者分类(或分期)、监测受试者中的肾功能、诊断慢性肾脏疾病(ckd)、监测受试者中ckd的进展或消退、诊断急性肾损伤(aki)、监测aki的进展或消退、评估受试者响应于组合物的肾功能、治疗具有ckd的受试者和治疗具有aki的受试者的方法。
本发明也提供了包含式i或式ii的化合物或其盐的试剂盒。
在另一个实施方案中,本发明的试剂盒包含式i或式ii的化合物或其盐以及关于测量生物样品中式i或式ii的化合物或其盐的水平的说明书。
在另一个实施方案中,所述试剂盒包括含有经标记的式i或式ii的化合物或其盐的内部标准品以及关于测量生物样品中式i或式ii的化合物或其盐的水平的说明书。
在另一个实施方案中,所述试剂盒包括式i或式ii的化合物或其盐、经标记的式i或式ii的化合物或其盐以及关于测量生物样品中式i或式ii的化合物的水平的说明书。
在一个实施方案中,本发明的试剂盒包含式i或式ii的化合物或其盐和基于在得自所述受试者的生物样品中检测到的化合物的水平来估计受试者中的gfr、确定受试者发生肾功能下降的倾向、根据肾功能的水平(或阶段)将受试者分类(或分期)、监测受试者中的肾功能、诊断慢性肾脏疾病(ckd)、监测受试者中ckd的进展或消退、诊断急性肾损伤(aki)、监测aki的进展或消退、评估受试者响应于组合物的肾功能、治疗具有ckd的受试者和治疗具有aki的受试者的说明书。
附图说明
图1显示了在碱性色谱条件下人血浆样品中的化合物a的lc/ms色谱图和波谱。
图2显示了具有放大的血浆样品中的化合物a的产物离子波谱(ms2)。
图3显示了在酸性色谱条件下人血浆样品(a)、人尿液样品(b)以及人血浆和尿的混合物(c)中的化合物a的lc/ms色谱图。
图4a和4b显示了在尿液样品中使用碰撞诱导的解离(cid)产生的具有放大的化合物a的产物离子波谱(ms2)。
图5a和5b显示了在尿液样品中使用高能碰撞诱导的解离(hcd)产生的具有放大的化合物a的产物离子波谱(ms2)。
图6a、6b、6c、6e、6d和6f显示了在尿液样品中的化合物a的ms3波谱:使用cid的m/z85(a)、115(b)、129(c)、159(d),和使用hcd的m/z85(e)、159(f)。
图7显示了早期洗脱峰的收集级分的浓缩残余物的1hnmr(700mhz),显示了亚砜。
图8显示了收集级分的浓缩残余物的lc/ms/ms色谱图和波谱,显示了在2.42和2.53分钟的亚砜和它们的对应ms2波谱。
图9显示了wittig产物的混合物的lc/ms色谱图,分别显示了亚砜作为在2.14、2.32、2.42和2.53分钟的痕量组分的存在,和在4.45和4.49分钟的受保护的反式和顺式硫代烯醇醚。
图10显示了收集级分的浓缩残余物的lc/ms色谱图和波谱,分别显示了具有m/z273.08049和273.08046的在2.42和2.53分钟的亚砜,和具有m/z257.08545的在4.45分钟的作为痕量组分的受保护的反式硫代烯醇醚。
图11显示了反式-dmtpa(2.54分钟,上部)、顺式和反式dmtpa的混合物(分别2.21和2.53分钟,中间)和它们的共注射(底部)的lc/ms色谱图。
图12显示了在尿液样品中的氘交换的化合物a的全扫描lc/ms色谱图和质谱图。
图13a和13b显示了在尿液样品中使用cid的具有放大的氘交换的化合物a的ms2波谱。
图14a和14b显示了在尿液样品中(a)具有放大(b)的使用cid的氘交换的化合物a的m/z160的ms3波谱。
图15a和15b显示了在尿液样品中(a)具有放大(b)的使用cid的氘交换的化合物a的m/z161的ms3波谱。
图16a、16b、16c、16d和16e显示了在尿液样品中使用cid的氘交换的化合物a的m/z130(a)、131(b)、86(c)、87(d)和116(e)的ms3波谱。
图17a、17b、17c和17d显示了在尿液样品中的氢化化合物a的lc/ms质谱图(a-b)和色谱图(d)。图c显示了天然化合物a(~2.5分钟)的提取离子色谱图,其在氢化以后不存在。
图18a和18b显示了在尿液样品中使用cid的具有放大的氢化化合物a的产物离子波谱(ms2)。
图19a和19b显示了在尿液样品中使用cid(a)或hcd(b)的氢化化合物a的m/z131的ms3波谱。
图20显示了在尿液样品中的氢化化合物a的氘化物质的lc/ms色谱图和质谱图。
图21a和21b显示了在尿液样品中使用cid的具有放大的氢化化合物a的氘化物质的产物离子波谱(ms2)。
图22显示了在尿液样品中使用cid的氢化化合物a的氘化物质的m/z132的ms3波谱。
图23显示了在尿液样品中使用cid的氢化化合物a的氘化物质的m/z133的ms3波谱。
图24显示了合成顺式和反式dmtpa的lc/ms色谱图和质谱图。
图25a、25b和25c显示了在尿液样品中的化合物a(2.53分钟)(a)、分别在2.20和2.53分钟的合成顺式和反式dmtpa(b)和它们的共注射(c)的lc-ms色谱图。
图26a和26b显示了使用cid的具有放大的合成反式dmtpa的产物离子波谱(ms2)。
图27a和27b显示了使用hcd的具有放大的合成反式dmtpa的产物离子波谱(ms2)。
图28a、28b、28c和28d显示了使用cid的合成反式dmtpa的m/z115(a)、129(b)、159(c)和85(d)的ms3波谱。
图29a和29b显示了使用hcd的合成反式dmtpa的m/z159(a)和85(b)的ms3波谱。
图30a和30b显示了合成反式dmtpa的氘化物质(a)和在尿液样品中的氘化物质化合物a(b)的全扫描质谱图。
图31a和31b显示了使用cid的具有放大的氘化合成反式dmtpa的产物离子波谱(ms2)。
图32显示了使用cid的具有放大的氘化合成反式dmtpa的m/z160的ms3波谱。
图33显示了使用cid的具有放大的氘化合成反式dmtpa的m/z161的ms3波谱。
图34显示了经纯化的wittig反应产物(受保护的顺式和反式硫代烯醇醚的混合物)的13cnmr谱。
图35显示了经纯化的wittig反应产物(受保护的顺式和反式硫代烯醇醚的混合物)的lc/ms色谱图和质谱图。
图36显示了使用cid的具有放大的经纯化的wittig反应产物(2.81分钟的rt)的产物离子波谱(ms2)。
图37显示了使用cid的具有放大的经纯化的wittig反应产物(2.98分钟的rt)的产物离子波谱(ms2)。
具体实施方式
定义
除非另外指出,否则本文中使用的以下术语定义如下:
本发明的化合物可以以盐的形式存在。任何合适的有机或无机盐都被包括在本发明中。在某些实施方案中,本发明的化合物的盐表示无毒的“药学上可接受的盐”。本文使用的短语“药学上可接受的”表示这样的化合物、物质、组合物和/或剂型:在合理的医学判断范围内,其适用于接触人类和动物的组织,而没有过度的毒性、刺激、变应性应答或其它问题或并发症、并与合理的收益/风险比相称。
本文中使用的“药学上可接受的盐”表示所公开的化合物的衍生物,其中母体化合物通过制备其酸式或碱式盐而被修饰。药学上可接受的盐形式包括药学上可接受的酸性的/阴离子的或碱性的/阳离子的盐。药学上可接受盐的例子包括、但不限于碱性残基如胺的无机酸盐或有机酸盐;酸性残基如羧酸的碱盐或有机盐;等。
例如,这样的盐包括乙酸盐、抗坏血酸盐、苯磺酸盐、苯甲酸盐、苄酸盐(benzylate)、碳酸氢盐、酒石酸氢盐、溴化物、依地酸钙、樟脑磺酸盐、碳酸盐、氯化物、柠檬酸盐、二盐酸盐、依地酸盐、乙二磺酸盐、乙烷二磺酸盐、依托酸盐、乙磺酸盐、富马酸盐、葡庚糖酸盐、葡萄糖酸盐、谷氨酸盐、乙醇酸盐、乙醇酰基对氨基苯胂酸盐、己基间苯二酚盐、哈胺(hydrabamine)、氢溴酸盐、盐酸盐、羟基马来酸盐、羟基萘甲酸盐、碘化物、羟乙基磺酸盐、乳酸盐、乳糖酸盐、苹果酸盐、马来酸盐、扁桃酸盐、甲磺酸盐(methanesulfonate)、甲磺酸盐(mesylate)、甲基溴化物、甲基硝酸盐、甲基硫酸盐、粘酸盐、萘磺酸盐、硝酸盐、草酸盐、扑酸盐、泛酸盐、苯基乙酸盐、磷酸盐/二磷酸盐、聚半乳糖醛酸盐、丙酸盐、水杨酸盐、硬脂酸盐、次醋酸盐、琥珀酸盐、磺酰胺、硫酸盐、鞣酸盐、酒石酸盐、8-氯茶碱盐、甲苯磺酸盐、三乙基碘、铵、苄星青霉素、氯普鲁卡因、colline、二乙醇胺、乙二胺、葡甲胺和普鲁卡因盐。可以与来自金属(如铝、钙、锂、镁、钾、钠、锌等)的阳离子形成其它药学上可接受的盐(也参见pharmaceuticalsalts,birge,s.m.等人,j.pharm.sci.,(1977),66,1-19)。
通过常规化学方法可以从含有碱性或酸性部分的母体化合物合成本发明的药学上可接受的盐。通常,可以如下制备这样的盐:在水中或在有机稀释剂如乙醚、乙酸乙酯、乙醇、异丙醇或乙腈或其混合物中,使这些化合物的游离酸或碱形式与足够量的适当碱或酸反应。
除上面提到的那些以外的酸的盐(例如三氟乙酸盐),其例如可用于纯化或分离本发明的化合物,也构成本发明的一部分。
通过异构体特异性合成或从异构混合物中拆分,可以将本发明的化合物制备为单一异构体。常规拆分技术包括使用光学活性的酸形成异构对的每种异构体的游离碱的盐(随后分步结晶和再生游离碱),使用光学活性的胺形成异构对的每种异构体的酸形式的盐(随后分步结晶和再生游离酸),使用光学纯的酸、胺或醇形成异构对的每种异构体的酯或酰胺(随后色谱分离并除去手性助剂),或使用各种众所周知的色谱方法拆分起始原料或终产物的异构体混合物。
当通过结构命名或描绘所公开的化合物的立体化学时,命名或描绘的立体异构体相对于其它立体异构体是按重量计至少60%、70%、80%、90%、95%、99%或99.9%纯的。当通过结构命名或描绘单一对映异构体时,所描绘或命名的对映异构体是按重量计至少60%、70%、80%、90%、95%、99%或99.9%光学纯的。按重量计的光学纯度百分比是对映异构体的重量与对映异构体的重量加其光学异构体的重量之比。
当所公开的化合物通过结构命名或描绘而未指示立体化学并且该化合物具有至少一个手性中心时,应当理解,该名称或结构涵盖不含对应光学异构体的该化合物的一种对映异构体、化合物的外消旋混合物和相对于其相应光学异构体富含一种对映异构体的混合物。
当所公开的化合物通过结构命名或描绘而未指示立体化学并且其具有至少两个手性中心时,应当理解,该名称或结构涵盖不含其它非对映异构体的非对映异构体、不含其它非对映异构体对的非对映异构体对、非对映异构体的混合物、非对映体对的混合物、其中一种非对映异构体相对于其它非对映异构体富集的非对映异构体混合物和其中一种非对映体对相对于其它非对映体对富集的非对映体混合物。
当用至少一个立构中心的特定立体化学描绘具有一个或多个立构中心的化合物时,本发明还包括在对应的立构中心处具有相反立体化学的化合物和在对应的立构中心处没有特定立体化学的化合物。
“治疗”病症或疾病表示治愈以及改善该病症或疾病的至少一种症状。
本文中使用的术语”受试者”是指任何动物,但优选地是哺乳动物,例如人、猴、非人灵长类动物、大鼠、小鼠、牛、狗、猫、猪、马或兔。甚至更优选地,受试者是人。
本文中使用的“有效量”是指在受试者中引起期望的生物应答的活性化合物试剂的量。这样的应答包括正在治疗的疾病或障碍的症状的减轻。在这样的治疗方法中,本发明的化合物的有效量是约0.01mg/kg/天至约1000mg/kg/天或约0.1mg/kg/天至约100mg/kg/天。
本发明的化合物或一种或多种另外化合物的“水平”是指在样品中测量的化合物的绝对或相对量或浓度。
“样品”或“生物样品”是指从受试者分离的生物材料。生物样品可以含有适合用于检测期望的化合物的任何生物材料,并且可以包含来自受试者的细胞和/或非细胞材料。样品可以分离自任何合适的生物组织或流体,例如,肾组织、血液、血浆(血浆)、血清(血清)、尿、唾液或脑脊液(csf)。优选地,生物样品是血液、血浆、血清、唾液或尿。在另一个优选的实施方案中,生物样品是血清或血浆。
“参比水平”是指本发明的化合物或另外化合物的水平,该水平指示特定疾病状态、表型或其缺乏,以及疾病状态、表型或其缺乏的组合。“参比水平”可以是本发明的化合物或另外化合物的绝对或相对量或浓度,本发明的化合物或另外化合物的存在或不存在,本发明的化合物或另外化合物的量或浓度的范围,本发明的化合物或另外化合物的最小和/或最大量或浓度,本发明的化合物或另外化合物的平均量或浓度,和/或本发明的化合物或另外化合物中位量或浓度;此外,本发明的化合物与另外化合物的组合的“参比水平”也可以是两种或多种化合物相对于彼此的绝对或相对量或浓度之比。通过在一个或多个合适的受试者中测量本发明的化合物或期望的化合物的水平,可以确定本发明的化合物或另外化合物对于特定疾病状态、表型或其缺乏而言适当的参比水平。“阳性”参比水平是指指示特定疾病状态或表型的水平。“阴性”参比水平是指指示缺乏特定疾病状态或表型的水平。
本文中使用的“参考样品”表示含有参比水平的化合物的样品。例如,可以从没有特定疾病、疾病状态或表型(诸如ckd或急性肾损伤)的受试者获得参考样品。
“肾小球滤过率”或“gfr”是每单位时间从肾小球毛细血管过滤到肾小囊中的流体的量。gfr是肾功能的度量,其中等于或高于某个阈值的gfr指示正常的肾功能,低于阈值的gfr指示肾功能受到损害或受损。通常,高gfr值指示较好的肾功能,而低gfr指示肾功能受损(例如,慢性肾脏疾病、急性肾损伤)。
“测得的肾小球滤过率”或“mgfr”是指实际的肾小球滤过率,其使用使用过滤标志物诸如菊粉、碘酞酸盐或碘海醇来确定。mgfr是在临床环境中执行的,且是肾功能的最准确测量。
“估计的肾小球滤过率”或“egfr”是指实际的肾小球滤过率的计算估计。所计算的值可以是基于一种或多种化合物或生物标志物的水平,并且可以包括其它变量,诸如人口统计信息(例如年龄或性别)。一种当前的计算egfr的方法是基于血清肌酸酐浓度。用于计算egfr的其它当前方法单独地或与血清肌酸酐的量结合地使用半胱氨酸蛋白酶抑制剂c的量。当前使用的用于估计gfr的公式包括“慢性肾脏疾病流行病学协作”(ckd-epi)和“肾病饮食改进egfr”(mdrd)。通常,低egfr值与肾功能下降有关。
“尿白蛋白”是测量尿液中的白蛋白的量的试验,也用于检测肾脏疾病。
“血清肌酸酐”或“scr”表示血清中的肌酸酐的测量,通常用于估计gfr。
“血液尿素氮”或“bun”表示以尿素的形式测量血液中的氮的量。bun是用于测量肾功能的试验。
“慢性肾脏疾病”或“ckd”包括这样的病症:其损害肾脏,导致肾脏从体内除去废物的能力下降,从而导致体内废物的高水平并导致增加的患病风险和并发症(诸如高血压、贫血、差的营养健康和神经损伤)的发生。具有肾功能异常至少三个月的患者可以被诊断为具有ckd。由ckd引起的肾损伤是永久性的。
“急性肾损伤”或“aki”表示其中肾功能迅速丧失的病症。由aki引起的肾损伤可能是可逆的。
“慢性肾脏疾病阶段”或“ckd阶段”是指当前使用测量的或估计的肾小球滤过率(mgfr,egfr)评估的肾损伤程度。临床上,通常公认5个阶段的ckd,肾功能在第1阶段被认为是正常的(gfr>90),在第2阶段被最小程度地降低(gfr60-89),在第3a和3b阶段被中度降低(gfr30-59),在第4阶段(gfr15-29)被严重降低,以及非常严重的或末期的肾衰竭,也被称为在第5阶段的已确定的肾衰竭(gfr<15或透析时)。肾功能阶段可以用于表示存在任何时间量的肾损伤(即,由aki或ckd引起的肾损伤)。
通过参考以下详细描述和实施例,可以更全面地理解本发明,所述详细描述和实施例意图例证本发明的非限制性实施方案。
化合物和组合物
本发明提供了新颖的化合物、组合物、合成方法、检测方法以及它们在诊断方法和治疗方法中的用途。
在第1个实施方案中,本发明提供了由下式表示的化合物:
或其盐。
在某些实施方案中,在第1个实施方案中的式(i)的化合物由下式表示:
或其盐。
在某些实施方案中,在第1个实施方案中的式(i)或式(ii)的化合物由下式表示:
或其盐。
在某些实施方案中,在第1个实施方案中的式(i)或式(ii)的化合物由下式表示:
或其盐。
在一个实施方案中,式(ii)、(iii)或(iv)的化合物或其盐是至少60%光学纯的、至少70%光学纯的、至少80%光学纯的、至少90%光学纯的、至少95%光学纯的或至少99%光学纯的。
在不同的实施方案中,本文描述的本发明的化合物(例如,由式(i)、(ii)、(iii)或(iv)表示的化合物或其盐)基本上不含有杂质。
在不同的实施方案中,本文描述的本发明的化合物(例如,由式(i)、(ii)、(iii)或(iv)表示的化合物或其盐)是至少60%纯的、至少70%纯的、至少80%纯的、至少90%纯的、至少95%纯的或至少99%纯的。
在某些实施方案中,本文描述的本发明的化合物(例如,由式(i)、(ii)、(iii)或(iv)表示的化合物或其盐)被同位素标记。在一个实施方案中,式(i)、(ii)、(iii)或(iv)的化合物或其盐被放射性地标记,诸如用氚(3h)或碳14(14c)。在另一个实施方案中,用氘、碳13(13c)、氧17(17o)、氧18(18o)、硫33(33s)、硫34(34s)或任意其它已知的硫同位素或它们的组合标记式(i)、(ii)、(iii)或(iv)的化合物或其盐。可以使用任意合适的同位素标记本发明的化合物的方法。
在某些实施方案中,本发明的同位素标记的化合物由下式表示:
或其药学上可接受的盐,其中*指示13c。
上述的化合物,诸如式(i)、(ii)、(iii)、(iv)、(l1)、(l2)、(l3)、(l4)、(l5)、(l6)、(l7)或(l8)的化合物或其盐(例如,药学上可接受的盐)可以用在本文所述的任何方法中。例如,本发明的化合物可以用于评估受试者的肾功能,计算受试者的肾小球滤过率的估计值,监测受试者以检测肾功能的改变(例如,功能的下降可能指示急性肾损伤或早期ckd),根据肾功能的程度将受试者分类(例如正常、轻度降低、中度降低、严重降低、终末期肾衰竭),和相对于未确诊ckd的对照受试者区分患有ckd的受试者。此外,所述化合物可以用于监测肾脏功能随时间或响应于药物治疗、疾病(例如,ii型糖尿病)或生活方式干预(例如,饮食、锻炼)的变化,和鉴别或排除受试者作为药物治疗和/或肾脏移植的合适候选人。
本发明还包括特异性地结合本文所述的化合物(例如式(i)、(ii)、(iii)或(iv)的化合物或其盐(例如,药学上可接受的盐))的抗体或抗体片段。用于产生特异性地结合小分子的抗体的方法是本领域已知的。还包括抗体衍生物,诸如包含上述抗体的vh和vl序列的多肽。在某些实施方案中,所述多肽是融合蛋白。本发明还包括用于产生本文描述的抗体或抗体片段和抗体衍生物的细胞。在一个实施方案中,所述细胞是真核细胞。
方法
在第2个实施方案中,本发明提供了用于确定受试者中本发明的化合物(例如,式(i)、(ii)、(iii)或(iv)的化合物或其盐)的水平的方法,所述方法包括:(1)从得自所述受试者的生物样品制备分析样品;和(2)确定所述化合物的水平。
为了确定在样品中的本发明的化合物(例如,式(i)、(ii)、(iii)或(iv)的化合物或其盐)的水平,分析了生物样品。分析样品的方法包括色谱法(例如,hplc、气相色谱法、液相色谱法)、质谱法(例如,ms、ms-ms)、酶联免疫吸附测定(elisa)、抗体连接、免疫印迹法、免疫组织化学(ihc)、其它免疫化学技术和它们的组合。
在一种示例性方法中,在得自所述受试者的生物样品中确定式(i)的化合物或其盐的水平包括使用质谱法确定式(i)的化合物的水平
在一个实施例中,可以在质谱法之前对生物样品进行液相色谱法(lc)。lc方法可以包括例如高效lc(hplc)或超高效lc(uhplc或uplc)。在某些实施例中,可以使用反相柱色谱系统、亲水相互作用色谱法(hilic)、离子交换色谱法或混合相柱色谱系统进行hplc或uplc。
使用包括离子源的质谱仪进行质谱法,所述离子源用于电离分级分离的样品和创建带电荷的分子用于进一步分析。通过例如加热的电喷射电离(hesi-ii),可以进行样品的电离。可以以正或负模式电离样品。
在已经电离样品以后,可以分析带正电荷或带负电荷的离子以确定质荷比。用于确定质荷比的示例性合适分析仪包括四极分析仪、离子阱分析仪、傅里叶变换质谱法(ftms)分析仪和飞行时间分析仪。
在某些实施方案中,通过质谱法确定样品中化合物a的水平的方法包括检测一种或多种离子,所述离子选自具有约177、159、133、131、129、115、113、111、101、100、99、89、85、83、75、73、57.03、57.00、56、97、87、74、84和67的质荷比(m/z)的离子。
分析结果可能包括由串联质谱法产生的数据。在某些实施例中,串联质谱法可以是准确质量串联质谱法。例如,准确质量串联质谱法可以使用四极飞行时间(q-tof)分析仪。在其它实施例中,串联质谱法可以是ftms。串联质谱法允许创建表示复杂混合物中的化学组分的母子关系的数据结构。该关系可以由树状结构表示,该树状结构示出了母离子和子离子彼此之间的关系,其中子离子代表母离子的子组分。
在某些实施方案中,可以使用本发明的化合物(例如,式(i)、(ii)、(iii)或(iv)的化合物或其盐(例如,药学上可接受的盐))的水平,例如,评估受试者的肾功能,计算受试者的肾小球滤过率的估计值,监测受试者以检测肾功能的改变(例如,功能的下降可能指示急性肾损伤或早期ckd),根据肾功能的程度将受试者分类(例如正常、轻度降低、中度降低、严重降低、终末期肾衰竭),和相对于未确诊ckd的对照受试者区分患有ckd的受试者。此外,所述化合物可以用于监测肾脏功能随时间或响应于药物治疗、疾病(例如,ii型糖尿病)或生活方式干预(例如,饮食、锻炼)的变化,和鉴别或排除受试者作为药物治疗和/或肾脏移植的合适候选人。
在一种示例性方法中,评估受试者的肾功能包括在得自所述受试者的生物样品中确定式(i)的化合物或其盐的水平:
其中与参比水平相比升高的生物样品中的化合物水平指示受试者中下降的肾功能。
在第3个实施方案中,本发明提供了用于计算患者中的估计的肾小球滤过率(egfr)的方法,所述方法包括下述步骤:(1)在得自所述受试者的生物样品中确定式(i)的化合物的水平或其盐;和(2)使用利用化合物的测量水平的算法计算egfr,其中所述算法使用利用外源性过滤标志物测得的gfr来开发。任何合适的方法可以用于确定所述化合物的水平。在一个实施方案中,使用色谱法、质谱法、酶联免疫吸附测定(elisa)、抗体连接、免疫印迹法、免疫组织化学(ihc)、其它免疫化学方法或它们的组合来确定所述化合物的水平。
在某些实施方案中,使用与肾功能评估有关的一种或多种另外化合物来计算egfr。在一个实施方案中,所述一种或多种另外化合物选自假尿苷、n-乙酰基苏氨酸、2-c-吡喃型甘露糖基色氨酸、n-乙酰基丝氨酸、n-乙酰基丙氨酸、n6-氨甲酰基苏氨酰基腺苷、4-乙酰氨基丁酸酯、赤藓醇、肌-肌醇、赤酮酸盐、尿素、阿糖醇、n2,n2-二甲基鸟苷、n1-甲基腺苷、3-甲基戊二酰基肉碱、s-腺苷基高半胱氨酸、n-乙酰基甲硫氨酸、n6-乙酰基赖氨酸、犬尿氨酸、阿拉伯糖酸盐、琥珀酰肉碱、核糖、木糖酸盐、n-甲酰基甲硫氨酸、o-甲基儿茶酚硫酸盐、2-甲基丁酰基肉碱、苯基乙酰基谷氨酰胺、n2,n5-二乙酰基鸟氨酸、色氨酸、肌酸酐、尿酸盐、3-吲哚氧基硫酸盐、对甲酚硫酸盐和n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)。在另一个实施方案中,所述egfr算法进一步利用血清半胱氨酸蛋白酶抑制剂c水平。在另一个实施方案中,所述egfr算法进一步利用一种或多种选自年龄、性别和种族的人口统计参数。
在第4个实施方案中,对于在第2个和第3个实施方案中描述的方法,式(i)的化合物由式(ii)表示:
或其盐。
在第5个实施方案中,对于在第2个和第3个实施方案中描述的方法,式(i)或式(ii)的化合物由式(iii)表示:
或其盐。
在第6个实施方案中,对于在第2个和第3个实施方案中描述的方法,式(i)或式(ii)的化合物由式(iv)表示:
或其盐。
在第7个实施方案中,对于在第2个至第6个实施方案中描述的方法,通过串联的液相色谱法-质谱法(lc-ms/ms)确定所述化合物的水平。
在第8个实施方案中,在第2个至第7个实施方案中描述的方法进一步包括在数学模型中使用所确定的化合物水平以评估肾功能。
在第9个实施方案中,在第2个至第8个实施方案中描述的方法进一步包括分析所述生物样品以确定与肾功能评估有关的一种或多种另外化合物的水平。所述一种或多种化合物可以选自下述化合物:假尿苷、n-乙酰基苏氨酸、2-c-吡喃型甘露糖基色氨酸、n-乙酰基丝氨酸、n-乙酰基丙氨酸、n6-氨甲酰基苏氨酰基腺苷、4-乙酰氨基丁酸酯、赤藓醇、肌-肌醇、赤酮酸盐、尿素、阿糖醇、n2,n2-二甲基鸟苷、n1-甲基腺苷、3-甲基戊二酰基肉碱、s-腺苷基高半胱氨酸、n-乙酰基甲硫氨酸、n6-乙酰基赖氨酸、犬尿氨酸、阿拉伯糖酸盐、琥珀酰肉碱、核糖、木糖酸盐、n-甲酰基甲硫氨酸、o-甲基儿茶酚硫酸盐、2-甲基丁酰基肉碱、苯基乙酰基谷氨酰胺、n2,n5-二乙酰基鸟氨酸、色氨酸、肌酸酐、尿酸盐、3-吲哚氧基硫酸盐和对甲酚硫酸盐和n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)、和它们的组合。在一个实施方案中,所述另外化合物选自假尿苷、n-乙酰基苏氨酸、色氨酸、苯基乙酰基谷氨酰胺、2-c-吡喃型甘露糖基色氨酸、犬尿氨酸、肌-肌醇、n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)和肌酸酐。在另一个实施方案中,所述另外化合物选自假尿苷、n-乙酰基苏氨酸、色氨酸、苯基乙酰基谷氨酰胺和肌酸酐。
在某些实施方案中,在第9个实施方案所描述的方法中确定2、3、4、5、6、7、8、9、10、11、12或更多种另外化合物的水平。
在某些实施方案中,第9个实施方案所描述的方法进一步包括分析所述生物样品以确定假尿苷、n-乙酰基苏氨酸、色氨酸和苯基乙酰基谷氨酰胺的水平。
在某些实施方案中,第9个实施方案所描述的方法进一步包括分析所述生物样品以确定假尿苷、n-乙酰基苏氨酸、色氨酸、苯基乙酰基谷氨酰胺和肌酸酐的水平。
确定本发明的化合物和一种或多种另外化合物的水平可以在所述方法中允许更大的灵敏度和特异性。例如,两种化合物或在生物样品中的某些化合物的水平的比率的成对分析可以在评估肾功能和辅助肾功能评估中允许更大的灵敏度和特异性。
使用多种技术,包括简单对比(例如,手工对比),可以将式(i)、(ii)、(iii)或(iv)的化合物和/或一种或多种另外化合物的水平与肾功能参比水平进行对比。使用一种或多种统计分析(例如,t-检验、welch氏t-检验、wilcoxon氏秩和检验、关联分析、randomforest、t-评分、z-评分)或使用数学模型(例如,算法、统计模型),也可以将生物样品中的式(i)、(ii)、(iii)或(iv)的化合物和/或一种或多种另外化合物的水平与参比水平进行对比。例如,包含单一算法或多个算法的数学模型可以用于评估受试者的肾功能。
该方法的结果可以与可用于评估受试者的肾功能的其它方法和测量(或其结果)一起使用。例如,临床参数诸如bun、scr和/或尿白蛋白测量值;肾功能标志物,诸如β-2微球蛋白、β-trace、2-c-吡喃型甘露糖基色氨酸;以及患者信息例如ckd家族史或其它风险因素,可以与所述化合物一起使用。
在某些实施方案中,本发明的化合物和/或一种或多种另外化合物的水平可以用于数学或统计模型或公式中以给医师提供数字评分(“肾功能评分”),其指示肾功能的水平和/或受试者具有受损的肾功能(其可能指示aki或ckd)的可能性。所述评分是基于一种化合物和/或多种化合物的组合的临床上显著改变的参比水平。所述参比水平可以从算法中推导出,或者从受损的gfr的指标中计算出。用于确定受试者的肾功能评分的方法可以包括将样品中的一种或多种肾功能化合物的水平与一种或多种化合物的肾功能参比水平进行对比以确定受试者的肾功能评分。所述方法可以采用选自以下列表的任意数量的化合物:假尿苷、n-乙酰基苏氨酸、2-c-吡喃型甘露糖基色氨酸、n-乙酰基丝氨酸、n-乙酰基丙氨酸、n6-氨甲酰基苏氨酰基腺苷、4-乙酰氨基丁酸酯、赤藓醇、肌-肌醇、赤酮酸盐、尿素、阿糖醇、n2,n2-二甲基鸟苷、n1-甲基腺苷、3-甲基戊二酰基肉碱、s-腺苷基高半胱氨酸、n-乙酰基甲硫氨酸、n6-乙酰基赖氨酸、犬尿氨酸、阿拉伯糖酸盐、琥珀酰肉碱、核糖、木糖酸盐、n-甲酰基甲硫氨酸、o-甲基儿茶酚硫酸盐、2-甲基丁酰基肉碱、苯基乙酰基谷氨酰胺、n2,n5-二乙酰基鸟氨酸、色氨酸、肌酸酐、尿酸盐、3-吲哚氧基硫酸盐、对甲酚硫酸盐和n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)。通过任何方法,包括统计方法诸如回归分析,可以将多种化合物与肾脏功能相关联。
肾功能评分可以用于将受试者置于正常(即无肾功能损害)至轻度降低、中度降低、严重降低或终末期肾衰竭的肾功能范围内。肾功能评分的非限制性示例用途包括:评估肾功能;估计gfr;肾功能分类;发生ckd的易感性;发生aki的易感性;ckd的诊断和阶段;通过肾功能评分的定期确定和监测来监测ckd进展;监测肾移植接受者的肾功能状态;确定对治疗干预的应答;评价药物效力;和确定治疗剂和/或造影剂的耐受性。
在某些实施方案中,对于本文描述的本发明的方法,所述生物样品是血液、血浆、血清、唾液或尿。在一个实施方案中,所述生物样品是血浆。在另一个实施方案中,所述生物样品是血清。
在某些实施方案中,对于本文描述的本发明的方法,在用允许直接测量肾小球滤过率的试剂治疗之前,从受试者获得所述生物样品。
在某些实施方案中,对于本文描述的本发明的方法,所述受试者是人。
在某些实施方案中,对于本文描述的本发明的方法,所述受试者没有肾功能受损的症状或没有肾功能受损的已知风险因素。在其它实施方案中,所述受试者表现出发生慢性肾脏疾病的风险因素(例如,60岁以上、高血压、糖尿病、心血管疾病、ckd家族史)。在某些实施方案中,所述受试者先前已被诊断患有高血压或糖尿病。在某些实施方案中,所述受试者具有慢性肾脏疾病的家族史。在某些实施方案中,所述受试者具有肾功能受损的症状。在某些实施方案中,所述受试者是使用常规方法难以评估肾功能的受试者。
在某些实施方案中,所述受试者选自以下各项:肥胖,非常瘦,素食,慢性病和老年人。
在某些实施方案中,所述受试者是肾脏供体的候选者。
在某些实施方案中,所述受试者已经用或正在考虑用对肾脏有毒性作用的试剂治疗。在一个实施方案中,所述试剂是造影剂。在另一个实施方案中,所述试剂是用于治疗疾病或病症的治疗剂,诸如化学治疗剂。在另一个实施方案中,所述试剂是抗生素。
在某些实施方案中,所述方法可以用于监测具有ckd的受试者或被怀疑易患ckd的受试者(例如,由于ckd的家族病史、药物治疗、慢性病等处于风险中的受试者)的肾功能。在一个实施例中,本发明的化合物可以用于监测没有ckd的受试者的肾功能。例如,在被怀疑易患ckd的受试者中,本文所述的化合物可以用于监测ckd的发展。
试剂盒
本发明包括试剂盒,其用于测量生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的水平。
在某些实施方案中,本发明的试剂盒包含式(i)、(ii)、(iii)或(iv)的化合物或其盐。
在某些实施方案中,本发明的试剂盒包含式(i)、(ii)、(iii)或(iv)的化合物或其盐以及关于测量生物样品中的式(i)、(ii)、(iii)或(iv)的化合物的水平的说明书。
在某些实施方案中,本发明的试剂盒可以包含经标记的化合物(例如,内部标准品)或能够检测生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的试剂。
在某些实施方案中,本发明的试剂盒可以包含经标记的化合物(例如,内部标准品)或能够检测生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的试剂以及关于测量生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的水平的说明书。
在一个实施方案中,上述试剂盒中的内部标准品是经标记的式(i)、(ii)、(iii)或(iv)的化合物或其盐(例如,式(l1)、(l2)、(l3)、(l4)、(l5)、(l6)、(l7)或(l8)的化合物或其盐)。
在一个实施方案中,本发明的试剂盒包含未标记的式(i)、(ii)、(iii)或(iv)的化合物或其盐、经标记的式(i)、(ii)、(iii)或(iv)的化合物或其盐(例如,式(l1)、(l2)、(l3)、(l4)、(l5)、(l6)、(l7)或(l8)的化合物或其盐)(作为内部标准品)以及关于测量生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的水平的说明书。
在某些实施方案中,本发明的试剂盒可以包含经标记的化合物(例如,内部标准品,诸如式(l1)、(l2)、(l3)、(l4)、(l5)、(l6)、(l7)或(l8)的化合物或其盐)。在其它实施方案中,所述试剂盒可以包含能够检测生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的试剂和用于确定样品中的化合物的量的装置(例如,针对式(i)、(ii)、(iii)或(iv)的化合物或其盐的抗体)。
在某些实施方案中,使用本文描述的本发明的试剂盒,通过色谱法、质谱法、酶联免疫吸附测定(elisa)、抗体连接、免疫印迹法、免疫组织化学(ihc)、其它免疫化学方法或它们的组合,可以确定生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的量。在某些实施方案中,通过色谱法、质谱法或它们的组合,可以确定生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的量。在某些实施方案中,使用本发明的试剂盒,通过lc-ms可以确定生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的量。
所述试剂盒也可以包含,例如,缓冲剂、防腐剂或稳定剂。所述试剂盒还可以含有一种对照样品或一系列对照样品,可以对其测定并与试验样品进行对比。试剂盒的每个组分通常被包封在单独的容器中,并且所有各个容器是在单个包装内,伴有关于确定被测受试者是否正在遭受与有关化合物有关的障碍或者处于发生所述障碍的风险中的说明书。
在某些实施方案中,本发明的试剂盒可以包括说明书,它是关于评估或监测受试者中的肾功能,确定发生肾功能下降的倾向,根据肾功能水平对受试者分类,诊断或监测ckd,诊断或监测aki,或基于在得自所述受试者的生物样品中检测到的化合物的水平来估计受试者中的gfr。
在本发明的试剂盒的某些实施方案中,式(i)、(ii)、(iii)或(iv)的化合物被同位素标记。在一个实施方案中,式(i)、(ii)、(iii)或(iv)的化合物被放射性地标记,例如,用氚(3h)或碳14(14c)。在另一个实施方案中,式(i)、(ii)、(iii)或(iv)的化合物被氘化,用碳13(13c)、氧17(17o)、氧18(18o)、硫33(33s)、硫34(34s)或任意其它已知的硫同位素或它们的组合标记。在另一个实施方案中,所述经标记的式(i)、(ii)、(iii)或(iv)的化合物由上述的式(l1)、(l2)、(l3)、(l4)、(l5)、(l6)、(l7)或(l8)表示。在一个实施例中,所述经标记的式(i)、(ii)、(iii)或(iv)的化合物(例如,式(l1)、(l2)、(l3)、(l4)、(l5)、(l6)、(l7)或(l8)的化合物或其盐)可以用作内部标准品。
本发明也提供了试剂盒,其包含特异性地结合上述式(i)、(ii)、(iii)或(iv)的化合物或其盐(例如,药学上可接受的盐)的抗体或抗体片段。在某些实施方案中,本发明的试剂盒包含抗体衍生物,诸如包含上述抗体的vh和vl序列的多肽。在一个实施方案中,所述多肽是融合蛋白。
在某些实施方案中,本发明的试剂盒包含特异性地结合式(i)、(ii)、(iii)或(iv)的化合物或其盐(例如,药学上可接受的盐)的抗体或抗体片段以及关于测量生物样品中的式(i)、(ii)、(iii)或(iv)的化合物或其盐的水平的说明书。
在某些实施方案中,本发明的试剂盒包含上述的抗体、抗体片段或抗体衍生物以及说明书,它是关于评估或监测肾功能,估计gfr,确定发生肾功能下降的倾向,根据肾功能水平对受试者分类,诊断或监测慢性肾脏疾病(ckd),或基于在得自所述受试者的生物样品中检测到的化合物的水平来诊断或监测受试者中的急性肾损伤(aki)。
在某些实施方案中,上述试剂盒包含一种或多种另外的生物标志物,其中所述一种或多种另外的生物标志物与肾功能的评估有关。本文描述的任何化合物可以用在本发明的试剂盒中。
在不同的实施方案中,所述一种或多种另外化合物选自假尿苷、n-乙酰基苏氨酸、2-c-吡喃型甘露糖基色氨酸、n-乙酰基丝氨酸、n-乙酰基丙氨酸、n6-氨甲酰基苏氨酰基腺苷、4-乙酰氨基丁酸酯、赤藓醇、肌-肌醇、赤酮酸盐、尿素、阿糖醇、n2,n2-二甲基鸟苷、n1-甲基腺苷、3-甲基戊二酰基肉碱、s-腺苷基高半胱氨酸、n-乙酰基甲硫氨酸、n6-乙酰基赖氨酸、犬尿氨酸、阿拉伯糖酸盐、琥珀酰肉碱、核糖、木糖酸盐、n-甲酰基甲硫氨酸、o-甲基儿茶酚硫酸盐、2-甲基丁酰基肉碱、苯基乙酰基谷氨酰胺、n2,n5-二乙酰基鸟氨酸、色氨酸、肌酸酐、尿酸盐、3-吲哚氧基硫酸盐、对甲酚硫酸盐和n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)、和它们的组合。在一个实施方案中,所述另外化合物选自假尿苷、n-乙酰基苏氨酸、色氨酸、苯基乙酰基谷氨酰胺、2-c-吡喃型甘露糖基色氨酸、犬尿氨酸、肌-肌醇、n,n,n-三甲基-l-丙氨酰基-l-脯氨酸甜菜碱(tmap)和肌酸酐。在另一个实施方案中,所述另外化合物选自假尿苷、n-乙酰基苏氨酸、色氨酸、苯基乙酰基谷氨酰胺和肌酸酐。
在某些实施方案中,本发明的试剂盒包含上述本发明的化合物(式(i)、(ii)、(iii)或(iv)的化合物或其盐)、假尿苷、n-乙酰基苏氨酸、色氨酸和苯基乙酰基谷氨酰胺。
在某些实施方案中,本发明的试剂盒包含上述本发明的化合物(式(i)、(ii)、(iii)或(iv)的化合物或其盐)、假尿苷、n-乙酰基苏氨酸、色氨酸、苯基乙酰基谷氨酰胺和肌酸酐。
制备方法
可以参考以下参考文献中适当的合成方法,如march,advancedorganicchemistry,第3版,johnwiley&sons,1985或wutsgreene’sprotectivegroupsinorganicsynthesis,第5版,johnwiley&sons2014和richardlarock,comprehensiveorganictransformations,第4版,vchpublishersinc,1989中所述。
在第一个实施方案中,通过包括下述步骤的方法,可以制备式(i)或式(ii)的化合物:
a)使式(v)的化合物或其盐
与wittig试剂诸如r’3p=chsme(也可以由它的共振结构r’3p+c-hsme表示)(其中r’是ph或吸电子基团)反应,以形成式(va)的化合物或其盐;
和
b)将式(va)的化合物或其盐去保护以形成式(i)的化合物或其盐。
在某些实施方案中,第一个实施方案的方法进一步包括纯化式(va)的化合物以产生式(vb)的化合物;
随后将式(vb)的化合物去保护以形成式(ii)的化合物或其盐。
在某些实施方案中,第一个实施方案的方法进一步包括纯化式(i)的化合物或其盐以分离反式-异构体和顺式-异构体从而产生式(ii)的化合物或其盐。
在第二个实施方案中,本发明提供了制备式(iii)的化合物的方法,所述方法包括下述步骤:
a)使式(vi)的化合物或其盐
与wittig试剂r’3p=chsme(其中r’是ph或吸电子基团)反应,以形成式(via)的化合物或其盐;
b)纯化(via)的化合物或其盐以产生式(vib)的化合物或其盐;
和
c)将式(vib)的化合物或其盐去保护以形成式(iii)的化合物或其盐。
可替换地,在第三个实施方案中,通过包括下述步骤的方法,可以制备式(iii)的化合物或其盐:
a)使式(vi)的化合物或其盐
与wittig试剂r’3p=chsme(其中r’是ph或吸电子基团)反应,以形成式(via)的化合物或其盐;
b)将式(via)的化合物或其盐去保护以形成式(vic)的化合物或其盐;
和
c)纯化式(vic)的化合物或其盐以产生式(iii)的化合物或其盐。
在某些实施方案中,可以在有酸存在下执行在第一个、第二个或第三个实施方案的方法中的去保护反应。可以使用任意合适的酸。示例性的酸包括、但不限于盐酸、乙酸、三氟乙酸、吡啶鎓对甲苯磺酸盐、对甲苯磺酸(p-tsoh)、甲酸、或氢形式的强或弱阳离子交换树脂。在某些实施方案中,所述酸是
在某些实施方案中,根据在第二个或第三个实施方案中描述的方法可以制备式(iv)的化合物或其盐,其中式(vi)的化合物用式(vi’)的化合物替换:
在某些实施方案中,使用同位素标记的起始原料和/或试剂可以制备同位素标记的式(i)、(ii)、(iii)或(iv)的化合物。在一个实施方案中,同位素标记的wittig试剂可以用在在第一个、第二个或第三个实施方案中描述的方法中,以制备同位素标记的式(i)、(ii)、(iii)或(iv)的化合物。在一个实施方案中,同位素标记的wittig试剂是ph3p=ch2scd3(也可以由它的共振结构ph3p+c-hscd3表示)。在另一个实施方案中,同位素标记的wittig试剂是ph3p=ch2s13cd3(也可以由它的共振结构ph3p+c-hs13cd3表示)。
在另一个实施方案中,同位素标记的2,3-亚环己基-赤糖醛酸可以用于制备同位素标记的式(i)、(ii)、(iii)或(iv)的化合物。在一个实施方案中,同位素标记的2,3-亚环己基-赤糖醛酸由下式表示:
其中*指示13c。
在第四个实施方案中,通过包括下述步骤的方法可以制备式(iii)的化合物:
a)使式(vii)的化合物或其盐:
与wittig试剂诸如r’3p=chsme反应,以形成式(viia)的化合物或其盐,
其中r’是ph或吸电子基团,且r1和r2各自独立地是甲硅烷基;
b)纯化式(viia)的化合物或其盐以产生式(viib)的化合物或其盐:
c)将式(viib)的化合物或其盐去保护以形成式(iii)的化合物。
可替换地,在第五个实施方案中,通过包括下述步骤的方法可以制备式(iii)的化合物:
a)使式(vii)的化合物或其盐
与wittig试剂诸如r’3p=chsme反应,以形成式(viia)的化合物或其盐,
其中r’是ph或吸电子基团,且r1和r2各自独立地是甲硅烷基;
b)将式(viia)的化合物或其盐去保护以形成式(viic)的化合物或其盐:
c)纯化式(viic)的化合物或其盐以产生式(iii)的化合物或其盐。
在某些实施方案中,根据在第四个或第五个实施方案中描述的方法可以制备式(iv)的化合物或其盐,其中式(vii)的化合物用式(vii’)的化合物替换:
在某些实施方案中,通过在第四个或第五个实施方案中描述的方法可以制备式(i)或(ii)的化合物,其中式(vii)的化合物用式(vii”)的化合物替换:
在第六个实施方案中,通过包括下述步骤的方法可以制备式(iii)的化合物或其盐:
a)使式(viii)的化合物或其盐:
与wittig试剂r’3pch2sme反应,以形成式(viiia)的化合物或其盐,
其中r’是ph或吸电子基团;r1和r2是甲硅烷基,且r3是叔丁基或r3是甲硅烷基;
b)纯化式(viiia)的化合物或其盐以产生式(viib)的化合物或其盐:
c)将式(viiib)的化合物去保护或其盐以形成式(iii)的化合物或其盐。
可替换地,在第七个实施方案中,通过包括下述步骤的方法可以制备式(iii)的化合物或其盐:
a)使式(viii)的化合物或其盐:
与wittig试剂r’3p=chsme反应,以形成式(viiia)的化合物或其盐,
其中r’是ph或吸电子基团;r1和r2是甲硅烷基,且r3是叔丁基或r3是甲硅烷基;
b)将式(viiia)的化合物或其盐去保护以形成式(viiic)的化合物或其盐:
c)纯化式(viiic)的化合物或其盐以产生式(iii)的化合物或其盐。
在某些实施方案中,根据在第六个或第七个实施方案中描述的方法可以制备式(iv)的化合物或其盐,其中式(viii)的化合物用式(viii’)的化合物替换:
在某些实施方案中,通过在第四个或第五个实施方案中描述的方法可以制备式(i)或(ii)的化合物,其中式(viii)的化合物用式(viii”)的化合物替换:
在某些实施方案中,在上述方法中(例如,在第一个、第二个、第三个、第四个、第五个、第六个或第七个实施方案中)的wittig试剂是甲硫基甲基三苯基鏻内鎓盐(ph3p=chsme(也可以由它的共振结构ph3p+c-hsme表示))。
在某些实施方案中,在有碱存在下执行在上述方法中(例如,在第一个、第二个、第三个、第四个、第五个、第六个或第七个实施方案中)的wittig反应。可以使用任意合适的碱。示例性的碱包括、但不限于双(三甲基硅烷基)氨基钠、双(三甲基硅烷基)氨基钾和正丁基锂。
在一个实施方案中,在有双(三甲基硅烷基)氨基钠存在下执行在上述方法中(例如,在第一个、第二个、第三个、第四个、第五个、第六个或第七个实施方案中)的wittig反应。
在某些实施方案中,在有机溶剂中执行上述方法(例如,在第一个、第二个、第三个、第四个、第五个、第六个或第七个实施方案中)的wittig反应。可以使用任意合适的有机溶剂或有机溶剂的混合物,其可以包括、但不限于四氢呋喃、乙腈、二氧杂环己烷、乙醚、甲基叔丁基醚、二氯甲烷等。例如,在四氢呋喃中执行wittig反应。
在另一个实施方案中,可以如下制备式(iii)的化合物或其盐:
使式(ix)的化合物或其盐
与甲硫醇或其盐反应,其中r1和r2各自独立地是甲硅烷基且r3是叔丁基或r3是甲硅烷基。
在第八个实施方案中,通过包括下述步骤的方法可以制备式(iii)的化合物:
a)使式(ix)的化合物与甲硫醇反应以形成式(viiia)的化合物或其盐:
b)纯化式(viiia)的化合物或其盐以产生式(viiib)的化合物或其盐:
c)将式(viiib)的化合物或其盐去保护以形成式(iii)的化合物或其盐。
在第九个实施方案中,通过包括下述步骤的方法可以制备式(iii)的化合物:
a)使式(ix)的化合物与甲硫醇反应以形成式(viiia)的化合物或其盐:
b)将式(viiia)的化合物或其盐去保护以形成式(viiib)的化合物或其盐:
c)纯化式(viiib)的化合物或其盐以产生式(iii)的化合物或其盐。
在某些实施方案中,通过在第七个或第八个实施方案中描述的方法可以制备式(iv)的化合物或其盐,其中式(ix)的化合物用式(ix’)的化合物替换:
在某些实施方案中,通过在第七个或第八个实施方案中描述的方法可以制备式(i)或(ii)的化合物或其盐,其中式(ix)的化合物用式(ix”)的化合物替换:
在某些实施方案中,可以在合成方案中使用炔烃硫氢化反应(即,在第八个或第九实施方案中与甲硫醇反应)。可以催化炔烃硫氢化,例如用紫外光照射、自由基引发剂诸如偶氮二异丁腈(aibn)、阳离子铑和铱复合物、钍和铀复合物、铑复合物、碳酸铯和/或金。
在某些实施方案中,上述甲硅烷基(例如在第四个、第五个、第六个、第七个、第八个或第九个实施方案中)包括、但不限于三烷基甲硅烷基(例如,二甲基异丙基甲硅烷基、二乙基异丙基甲硅烷基、二甲基己基甲硅烷基、三甲基甲硅烷基或三异丙基甲硅烷基)、三苄基甲硅烷基、三苯基甲硅烷基、2-降冰片基二甲基甲硅烷基、叔丁基二甲基甲硅烷基、叔丁基二苯基甲硅烷基、2-三甲基乙基甲硅烷基(teoc)或[2-(三甲基甲硅烷基)乙氧基]甲基)。在某些实施方案中,所述甲硅烷基是三烷基甲硅烷基。在某些实施方案中,所述甲硅烷基是三甲基甲硅烷基或三(三甲基甲硅烷基)甲硅烷基。
保护基r1、r2和r3的除去依赖于保护基的性质。在一个实施方案中,通过用去保护试剂(其为氟化物和/或酸)处理可以除去保护基。在某些实施方案中,所述去保护试剂包括、但不限于四正丁基氟化铵、三(二甲基氨基)锍二氟三甲基硅酸盐、氟化氢或其溶剂合物、氟化氢吡啶、四氟化硅、六氟硅酸、氟化铯、盐酸、乙酸、三氟乙酸、吡啶鎓对甲苯磺酸盐、对甲苯磺酸(p-tsoh)、甲酸或过碘酸。在某些实施方案中,所述去保护试剂是盐酸或四正丁基氟化铵(tbaf)。在某些实施方案中,所述去保护试剂是氟化氢-吡啶(hf-吡啶)。
在某些实施方案中,在上述方法中(例如,在第一个、第二个、第三个、第四个、第五个、第六个、第七个、第八个或第九个实施方案中)描述的纯化步骤可以通过任意合适的纯化方法执行。在某些实施方案中,所述纯化步骤导致对应的反式-异构体(双键的反式构型)与顺式-异构体的分离。在某些实施方案中,反式-异构体和顺式-异构体的分离可以通过色谱法或结晶实现。在某些实施方案中,反式-异构体和顺式-异构体的分离可以通过hplc实现。
实施例
材料和方法
试剂和仪器
质谱级甲酸(~98%)、hplc级三氟乙酸(≥99.0%)、活性炭载钯(10%)、(甲硫基甲基)三苯基氯化鏻、无水四氢呋喃、双(三甲基硅烷基)氨基钠(1.0m在四氢呋喃中)、2,3-亚环己基-l-赤糖醛酸、
色谱法
除非另外指出,否则使用配备二元溶剂管理器、冷藏样品管理器(设定在12℃)和柱管理器(设定在40℃)的watersacquityuplc系统进行用反相柱(watersacquity
色谱法:碱性条件
流动相a是6.5mm的碳酸氢铵水溶液,流动相b是6.5mm碳酸氢铵在甲醇/水(95:5)中的溶液。用0%流动相b的初始条件(将其保持1.50分钟)进行线性梯度洗脱。然后将流动相b在0.50分钟中增加至98%并维持0.90分钟。将流动相b在0.10分钟中恢复至0%为下一次注射平衡。总运行时间是4.00分钟。
色谱法:氢化产物的条件,酸性条件
流动相a是0.1%的甲酸水溶液,流动相b是0.1%的甲酸在乙腈中的溶液。用2%流动相b的初始条件(将其保持3.00分钟)进行线性梯度洗脱。然后将流动相b在0.40分钟中增加至98%并维持0.50分钟。将流动相b在0.10分钟中恢复至2%为下一次注射平衡。总运行时间是5.00分钟。
色谱法:氢化产物的条件,氘化的酸性条件
流动相a是0.1%的甲酸在氘化水中的溶液,流动相b是0.1%的甲酸在乙腈中的溶液。用2%流动相b的初始条件(将其在3.00分钟中增加至7%)进行线性梯度洗脱。然后将流动相b在0.40分钟中增加至98%并维持0.50分钟。将流动相b在0.10分钟中恢复至2%为下一次注射平衡。总运行时间是5.00分钟。
色谱法:wittig反应产物的条件
流动相a是0.1%的甲酸水溶液,流动相b是0.1%的甲酸在乙腈中的溶液。用38%流动相b的初始条件(将其在3.00分钟中增加至45%)进行线性梯度洗脱。然后将流动相b在0.40分钟中增加至98%并维持0.50分钟。将流动相b在0.10分钟中恢复至38%为下一次注射平衡。总运行时间是5.00分钟。
色谱法:双键测定的条件(亚砜)
流动相a是0.1%的甲酸水溶液,流动相b是0.1%的甲酸在乙腈中的溶液。用20%流动相b的初始条件(将其在3.00分钟中增加至30%)进行线性梯度洗脱。然后将流动相b在1.40分钟中增加至98%并维持0.50分钟。将流动相b在0.10分钟中恢复至20%为下一次注射平衡。总运行时间是6.00分钟。
质谱法
以负模式使用配备加热电喷射电离(hesi-ii)探针的thermoscientificorbitrapelite质谱仪。该仪器由orbitrapelitetm2.7和xcaliburtm2.2软件(后来升级为3.0)控制。将加热的电喷射源设置为加热器温度在430℃,鞘气在65,辅助气体流速在15,清扫气在0,离子喷射电压在3.25kv,毛细管温度在350℃,且s透镜rf水平在60%。使用30,000的分辨率收集具有在m/z100和600之间的质量范围的全扫描ftms波谱。将所有片段化实验设定成具有m/z50和200之间的扫描范围,15,000的分辨率,1.0的隔离宽度,0.250的活化q,和10.0ms的活化时间。在没有任何锁定质量的情况下采集和处理所有质谱数据,并且使用外部质量校准。
ms2实验的归一化碰撞能量对于m/z177.02(或氘交换为179.03)的cidms2实验为28.0ev,对于相应的hcdms2实验为100ev。对于m/z177.02/159.01(或氘交换为179.03/160.02和179.03/161.02)的cidms3实验,第一阶段和第二阶段片段化的归一化碰撞能量分别为26.0和25.0ev。对于m/z177.02/159.01的hcdms3实验,第一阶段(cid)和第二阶段(hcd)片段化的归一化碰撞能量分别为26.0和100ev。对于m/z177.02/129.02(或氘交换为179.03/130.03和179.03/131.03)的cidms3实验,第一阶段和第二阶段片段化的归一化碰撞能量分别为28.0和25.0ev。对于m/z177.02/115.02(或氘交换为179.03/116.03)的cidms3实验,第一阶段和第二阶段片段化的归一化碰撞能量分别为28.0和30.0ev。对于m/z177.02/85.03(或氘交换为179.03/86.04和179.03/87.04)的cidms3实验,第一阶段和第二阶段片段化的归一化碰撞能量分别为28.0和21.0ev。对于m/z177.02/85.03的hcdms3实验,第一阶段(cid)和第二阶段(hcd)片段化的归一化碰撞能量分别为28.0和200ev。
对于氢化产物,m/z179.04(或氘交换为181.05)的cidms2实验的归一化碰撞能量为28.0ev。对于m/z179.04/131.03(或氘交换为181.05/132.04和181.05/133.05)的cidms3实验,第一阶段和第二阶段片段化的归一化碰撞能量分别为28.0和25.0ev。对于m/z179.04/131.03的hcdms3实验,第一阶段(cid)和第二阶段(hcd)片段化的归一化碰撞能量分别为28.0和120ev。
对于wittig反应产物(顺式和反式保护的硫代烯醇醚的混合物),对于m/z257.09的cidms2实验,归一化的碰撞能量为26.0ev,扫描范围在m/z70和280之间。
对于亚砜并确定顺式和反式保护的硫代烯醇醚的双键构型,对于m/z273.08的cidms2实验,归一化碰撞能量为26.0ev,扫描范围在m/z75和280之间。
实施例1.血浆和尿代谢物化合物a的结构阐明
样品制备
在50ml塑料离心管中加入6ml合并的人血浆和30ml甲醇。将混合物涡旋约1分钟并在4℃在4000rpm离心15分钟。将上清液以每个孔500μl转移至96-孔板,并在40℃在温和的氮气流下干燥。将5个孔中的残余物依次在涡旋下在水(200ml)中重构(各1分钟)。然后将混合物转移至1.5mleppendorf试管并在室温在14,000rpm离心10分钟。然后将上清液转移至样品瓶用于lc/ms分析。将尿液样品在室温在14,000rpm离心10分钟,并将上清液(如果需要的话,用水稀释)转移至样品瓶用于lc/ms分析。关于化合物a在血浆和尿液样品中的保留时间对比,用1.0n盐酸将注射溶液调至ph~3.0。
化合物a的氢化
在20ml反应瓶中,加入4ml尿、30mg活性炭载钯(10%)和磁力搅拌棒。将瓶用氢气净化,并然后配备填充了氢气的气球。将反应混合物在室温搅拌过夜,并然后缓慢地穿过注射器式滤器(0.45μm)。将澄清滤液转移至样品瓶用于lc/ms分析。
结构阐明
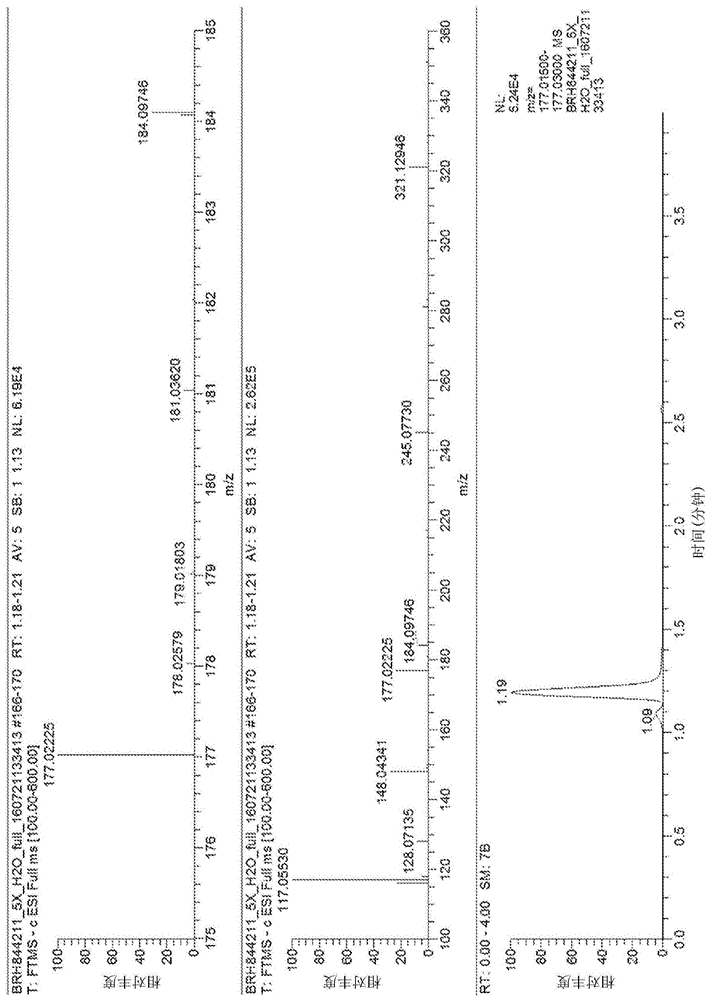
将orbitrapelite质谱仪用于采集高分辨率质谱图。质子化的假分子离子的分子式预先通过准确质量测量确定为c6h9o4s-,并且计算出单同位素质量为177.02270。通过使用血浆提取物优化色谱条件,开始当前的研究。在使用100%碳酸氢铵水溶液的碱性流动相条件下,化合物a仅轻微保留在反相柱上(1.19分钟,图1)。如在图2中所示,假分子离子的碰撞诱导的解离(cid)产生了许多在质量范围内的子离子。当使用酸性流动相条件时,化合物a在含0.1%甲酸的98%水和含0.1%甲酸的2%乙腈的相同反相柱上保留得更好(2.53分钟,图3a)。选择此方法用于进一步研究。
发现化合物a在血浆中以低水平存在。试验了尿液样品,与化合物a在血浆提取物中的保留时间匹配的峰在尿液中更强(图3b)。当注入尿液和血浆提取物的混合物时,尿液中的化合物与血浆中的化合物a共洗脱(图3c)。尿液中化合物的产物离子波谱(图4)也与血浆提取物中化合物a的产物离子波谱(图2)非常好地匹配。这些结果证实了化合物a以比在血浆中更高的浓度存在于尿中。使用尿液样品进行了进一步分析。
假分子离子的cid产物离子波谱产生了17个具有不同强度的子离子,如在图4中所示,它们是m/z159、133、131、129、115、113、111、101、100、99、89、85、83、75、73、57.03和57.00。通过高能碰撞诱导的解离(hcd)所产生的另一种产物离子波谱(图5)产生了另外三个子离子(m/z97、87和74)。m/z85、115、129和159离子(cidms3,图6a-d)和m/z85和159离子(hcdms3,图6e-f)的进一步片段化波谱检测到另外两个离子(m/z84和67),并建立了某些离子的关系。
基于这些片段及其形成途径的合理化,提出了化合物a的化学结构。提议的结构具有不确定的双键构型和不确定的立体化学,如下所示:
化合物a与5-甲硫基核糖(mtr)具有亲密的结构相似性,后者是从5’-甲硫基腺苷(mta)生物合成。
基于它与mtr和mta的结构相似性,指定化合物a的立体化学,并确定该通式由以下结构表示:
双键构型的确定
在中性流动相条件下将在实施例2方法1中合成的受保护的反式和顺式硫代烯醇醚的混合物的一部分半制备地色谱分离,并收集早期洗脱异构体的洗脱液和蒸发至干燥(称为hplc纯化的样品)。当通过1hnmr分析所得的残留物时(图7),波谱意外地与受保护的硫代烯醇醚的异构体(顺式或反式)不匹配。使用cosy、lc/ms和lc/ms2波谱的进一步分析表明,在早期洗脱异构体的纯化过程中形成了一对非对映异构的亚砜(2.42和2.53分钟)(lc/ms2,图8)。1hnmr谱显示烯族质子之间15hz的偶合常数,表明在非对映异构亚砜对中双键的构型是反式。由于亚砜是在纯化过程中从较早洗脱的受保护的硫代烯醇醚形成,因此确定受保护的硫代烯醇醚的早期洗脱异构体的双键构型是反式。
通过lc/ms分析在实施例2方法1中合成的受保护的反式和顺式硫代烯醇醚的混合物。如在图9中所示,通过lc/ms分别在4.45和4.49分钟检测到了受保护的反式和顺式硫代烯醇醚。尽管通过1hnmr在hplc纯化的样品中未观察到受保护的反式硫代烯醇醚,但是通过lc/ms分析在4.45分钟清楚地检测到它(图10),表明受保护的反式硫代烯醇醚不完全氧化为亚砜。受保护的反式硫代烯醇醚在hplc纯化的样品中仍存在,但相对于亚砜而言在低得多的水平。当在弱酸性条件下处理hplc纯化的样品时,实现保护基团从受保护的反式硫代烯醇醚的除去以形成反式-dmtpa,如通过lc/ms在2.54分钟检测到的。反式-dmtpa与顺式和反式dmtpa的混合物的晚期洗脱异构体共洗脱(图11)。因此,确定晚期洗脱dmtpa异构体具有反式双键构型。因此,确定化合物a具有反式双键构型,因为它也与晚期洗脱异构体共洗脱。具有指定的立体化学的化合物a的结构如下式(iii)所示。
如在方案1和2中所示,提出了从化合物a的去质子化的假分子离子的可能片段化途径。
方案1.可能片段化途径
方案2.可能片段化途径
通过氘交换进行的结构验证
通过氘交换实验验证了提议的化合物a的结构。简而言之,将色谱的流动相更改为氘化溶剂,并再次分析尿液提取物。获得全扫描质谱图,并检测到具有m/z179.03518离子(比c6h7d2o4s-的计算值偏离-0.4ppm)的新离子作为氘化化合物a的主要种类(图12),其与所提议的结构一致。显示了m/z179离子的产物离子波谱(ms2)(图13)和对应m/z160(图14)、161(图15)、130、131、86、87和116(图16a-e)离子的ms3波谱。在m/z161、135、131、117、103、102、87、77和59处检测到氘化片段,而将单个氘掺入m/z160、132、130、116、114、112、102、101、100、90、86、84、76和58。在m/z113、111、89、83、75、72和57处观察到没有掺入氘的片段。m/z160和161离子的检测指示hod和h2o分别从双氘化母离子的损失。m/z161离子的形成可能是由于在失去h2o之前在氧上的氘核与在碳上的质子交换而发生。所有这些离子都可以令人满意地合理化为最初提出的片段化途径(参见方案3-5),从而为提出的化合物a结构的有效性提供了令人信服的证据。
方案3.氘化类似物的可能片段化途径。
方案4.氘化类似物的可能片段化途径。
方案5.氘化类似物的可能片段化途径。
通过氢化进行的结构验证
所提出的结构含有碳-碳双键,并通过饱和实验进一步证实。对尿液样品进行了催化氢化,反应混合物的lc/ms分析表明与化合物a对应的峰(~2.5分钟)的消失和具有m/z179.03839的在3.17分钟处的峰的出现(图17),后者对应于c6h11o4s-的式(相对于179.03835的计算值偏离0.2ppm)。产物离子波谱(ms2)(图18)和m/z131离子的ms3波谱(cid和hcd)(图19)支持化合物a的饱和结构的鉴定,片段的合理化如在方案6中所示。
方案6.氢化类似物的可能片段化途径.
使用氘化流动相的lc/ms分析揭示,两个氘核掺入具有m/z181.05059的假分子离子中(图20),对应于c6h9d2o4s-的式(相对于181.05191的计算值偏离-1.77ppm)。氘化物质的产物离子波谱(图21)以及m/z132离子(图22)和m/z133离子(图23)的ms3波谱与预期的饱和产物一致。在方案7中列出了氘化物质的可能片段化途径。
方案7.氢化类似物的双氘化物质的可能片段化途径.
通过与合成标准品(参见实施例1)直接对比进一步证实了所提出的结构。通过lc/ms,由实施例2方法1制得的合成顺式和反式dmtpa混合物导致在2.19和2.53分钟的具有m/z177.02262的分子离子(偏离计算值-0.5ppm)的产物(图24)。选择通过方法1制备的在2.53分钟洗脱的产物(后来确定是反式dmtpa)用于与化合物a进一步对比。
合成反式dmtpa的保留时间(图25b)与化合物a在尿液中的保留时间(图25a)匹配,因为它们在色谱条件下共洗脱(图25c)。通过片段化模式和强度分布确定,通过cid(图26)和hcd(图27)得到的合成反式dmtpa的产物离子波谱(ms2)与那些化合物a(图4和5)相匹配。通过合成反式dmtpa的115、129、159和85子离子的cid进行进一步片段化,产生了ms3波谱(图28a-d)。159和85子离子的ms3波谱也由hcd产生(图29a-b)。合成反式dmtpa的所得ms3波谱与化合物a的ms3波谱基本上相同(图6)。此外,在氘交换后分析了合成反式dmtpa,并且得到的ms、ms2和ms3波谱也与化合物a的波谱非常好地匹配,如在图30-33中所示与图13-15相比。所有这些色谱和ms波谱数据都强烈地支持化合物a是反式dmtpa。
实施例2.(2r,3r)-2,3-二羟基-5-甲硫基-反式-4-戊烯酸(反式dmtpa)的合成
方法1.
wittig反应
在氩气氛下在25ml圆底烧瓶中将(甲硫基甲基)三苯基氯化鏻(500mg,1.39mmol,2.2当量)悬浮于四氢呋喃中。将混合物冷却至0℃,并在搅拌混合物的同时缓慢地加入双(三甲基硅烷基)氨基钠(1.0m在四氢呋喃中,1.39ml,1.39mmol,2.2当量)。将得到的混合物在0℃搅拌另外30分钟,并缓慢地加入2,3-亚环己基-l-赤糖醛酸(136mg在1.0ml四氢呋喃中,0.633mmol,1.0当量)。5分钟以后,将反应混合物温热至室温并搅拌过夜。将反应混合物转移至nah2po4水溶液(1.0m,25ml),并将得到的混合物(ph~6.0)用乙酸乙酯(2x40ml)萃取。将合并的有机萃取物干燥(mgso4),过滤,并蒸发至干燥以得到稠油(402mg)。将该粗提取物在硅胶柱(2.5x18cm)上色谱分离,用乙酸乙酯/甲醇(9:1)洗脱。将对应的级分合并,并浓缩至干燥以产生受保护的硫代烯醇醚的e/z(~5:4)异构体的混合物,作为淡棕色蜡样物质(75mg,46%)。1hnmr(500mhz,cd3od)δ6.57(1h,dd,j=14.9,0.8hz,h-5e),6.37(0.6h,dd,j=9.7,1.0hz,h-5z),5.49(0.6h,dd,j=9.6,8.7hz,h-4,z),5.26(1h,dd,j=14.9,8.0hz,h-4,e),5.23(0.8h,ddd,j=8.7,7.2,1.0hz,h-3,z),4.95(1h,overlapdoh,ddd,j=8.3,7.3,1.0hz,h-3,e),4.67(1h,d,j=7.3hz,h-2,e),4.66(0.9h,d,j=7.3hz,h-2,z),2.32(2h,s,ch3),2.26(3h,s,ch3),1.35-1.95(m,环质子)。13cnmr(125mhz,cd3od)δ173.2,173.1,135.1,132.6,123.8,119.9,112.7,112.5,79.5,78.4,77.8,75.6,37.9,36.01,35.96,26.4,25.2,25.0,17.4,14.3。
通过1h、13c和1h-1hcosynmr波谱和质谱分析表征了结构。经纯化的wittig反应产物(受保护的顺式和反式硫代烯醇醚的混合物)的13cnmr谱显示在图34中。经纯化的wittig反应产物的lc/ms色谱图和质谱图显示在图35中。具有2.81分钟或2.98分钟的保留时间的经纯化的wittig反应产物的产物离子波谱(ms2)分别显示在图36和37中。
去保护
在20ml闪烁瓶中,加入受保护的顺式和反式硫代烯醇醚的混合物(2.0mg在0.50ml乙腈中)、
通过将起始原料溶解在0.1%的甲酸水溶液中进行第二种去保护方法。在lc/ms分析之前,将溶液在室温保持1小时。
wittig反应产物(受保护的顺式和反式硫代烯醇醚的混合物)的hplc纯化
使用配备二元溶剂泵单元、冷藏自动采样器(设定在4℃)和柱加热器(设定在50℃)的agilent1290infinityuhplc系统进行用反相柱(waters
方法2.下面介绍了第二种合成方案的一般策略。受保护的l-赤糖醛酸可用作起始分子。可以使用任何合适的羟基保护基(p.g.m.wuts,greene’sprotectivegroupsinorganicsynthesis,wiley-interscience,newyork,2014)。在一个实施方案中,保护基r1和r2可以是甲硅烷基,诸如三烷基甲硅烷基(例如三甲基甲硅烷基)。wittig试剂可以是任何盐,其中xˉ可以是clˉ、brˉ、iˉ等。保护基r1和r2的除去取决于保护基的性质。在一个实施方案中,通过用氟化物处理可以除去保护基。
在该一般策略的另一个实施例中,起始原料可以是受保护的l-赤糖醛酸。羧基保护基r3可以是叔丁基或甲硅烷基(例如三(三甲基甲硅烷基)甲硅烷基)。保护基r1、r2和r3的除去取决于保护基的性质。在一个实施方案中,通过用氟化物和/或酸处理可以除去保护基。
方法3.下面介绍了第三种合成方案的一般策略。(2r,3r)-2,3-二羟基-4-戊炔酸或其保护形式可以用作起始分子。可以使用任何合适的羟基和羧基保护基(p.g.m.wuts,greene’sprotectivegroupsinorganicsynthesis,wiley-interscience,newyork,2014)。在一个实施方案中,保护基r1和r2可以是甲硅烷基,诸如三烷基甲硅烷基(例如三甲基甲硅烷基),且羧基保护基r3可以是叔丁基或甲硅烷基(例如三(三甲基甲硅烷基)甲硅烷基)。在一个实施方案中,在合成方案中可以使用炔烃硫氢化反应。可以催化炔烃硫氢化,例如,用紫外光照射、自由基引发剂诸如偶氮二异丁腈(aibn)、阳离子的铑和铱复合物、钍和铀复合物、铑复合物、碳酸铯和/或金。保护基r1、r2和r3的除去取决于保护基的性质。在一个实施方案中,通过用氟化物和/或酸处理可以除去保护基。
实施例3.化合物a的lc-ms/ms测量
使用配备二元溶剂管理器、冷藏样品管理器(设定在12℃)和柱管理器(设定在40℃)的watersacquityuplc系统进行用反相柱(watersacquity
使用配备有以负模式运行的加热电喷射电离(hesi-ii)探针的thermoscientificorbitrapelite质谱仪进行质谱分析。以高分辨率测量所有质量,但为了清楚起见简化了报告。该仪器由orbitrapelitetm2.7和xcaliburtm2.2软件(后来升级为3.0)控制。将加热的电喷射源设定为加热器温度在430℃,鞘气在65,辅助气体流速在15,清扫气在0,离子喷射电压在3.25kv,毛细管温度在350℃,且s透镜rf水平在60%。使用30,000的分辨率收集具有在m/z100和600之间的质量范围的全扫描ftms波谱。将所有片段化实验设定成具有m/z50和200之间的扫描范围,15,000的分辨率,1.0的隔离宽度,0.250的活化q,和10.0ms的活化时间。在没有任何锁定质量的情况下采集和处理所有质谱数据,并且使用外部质量校准。
ms2实验的归一化碰撞能量对于m/z177.02的cidms2实验为28.0ev,对于相应的hcdms2实验为100ev。具有约177.02的m/z的母离子的片段化产生了具有约159、133、131、129、115、113、111、101、100、99、89、85、83、75、73、57.03、57.00和56的m/z的子离子。hcd产生了具有约97、87和74的m/z的另外三种子离子。对于具有m/z177.02/159.01的cidms3实验,第一阶段和第二阶段片段化的归一化碰撞能量分别为26.0和25.0ev。对于m/z177.02/159.01的hcdms3实验,第一(cid)和第二(hcd)阶段片段化的归一化碰撞能量分别为26.0和100ev。对于m/z177.02/129.02的cidms3实验,第一和第二阶段片段化的归一化碰撞能量分别为28.0和25.0ev。对于m/z177.02/115.02的cidms3实验,第一和第二阶段片段化的归一化碰撞能量分别为28.0和30.0ev。对于m/z177.02/85.03的cidms3实验,第一和第二阶段片段化的归一化碰撞能量分别为28.0和21.0ev。对于m/z177.02/85.03的hcdms3实验,第一(cid)和第二(hcd)阶段片段化的归一化碰撞能量分别为28.0和200ev。约177.02/159.01、177.02/129.02、177.02/115.02和177.02/85.03的m/z的ms3片段化产生了具有约84和67的m/z的两种另外离子。
- 还没有人留言评论。精彩留言会获得点赞!