用于诊断肺癌的组合物、方法和试剂盒与流程
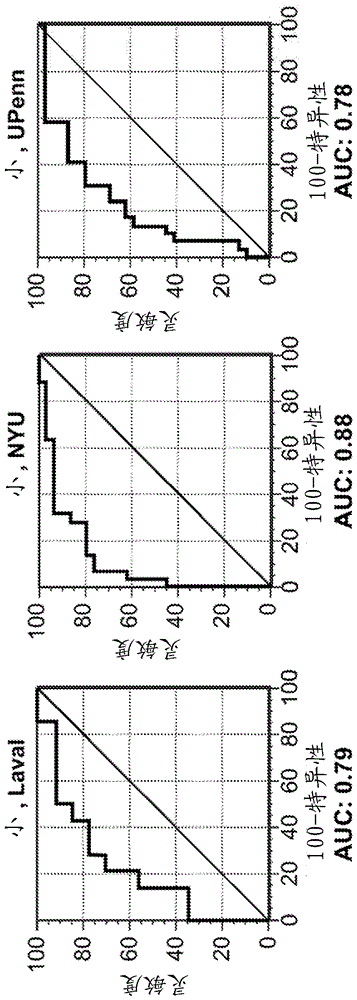
相关申请本申请是于2017年10月18日提交的美国申请号15/786,924的继续申请,其为于2016年2月23日提交的美国申请号15/051,153的部分继续申请,其为于2013年2月25日提交的美国申请号13/775,494,现在的美国专利号9,304,137的继续申请,其为于2012年12月21日提交的美国申请号13/724,823,现在的美国专利号9,201,044的部分继续申请,其要求以下的优先权和权益:于2011年12月21日提交的美国申请号61/578,712、于2012年1月24日提交的美国申请号61/589,920、于2012年7月27日提交的美国申请号61/676,859、以及于2012年11月12日提交的美国申请号61/725,153,所述申请各自的内容整体引入本文作为参考。引入作为参考的序列表于2015年2月27日创建且大小为14kb的命名为“idia-005_x02us_sequencelisting_st25.txt”的文本文件的内容整体引入本文作为参考。背景肺部状况且特别是肺癌呈现了重大的诊断挑战。在许多无症状患者中,放射学筛查例如计算机断层摄影(ct)扫描是诊断范例中的第一步。肺结节(pn)或不确定结节位于肺部,且经常在筛查高危患者过程中或偶然地发现。由于获得卫生保健的患者数量渐增、筛查技术的快速采用和人口老龄化,鉴定的pn数目预期上升。据估计,美国每年鉴定了超过300万例pn。尽管大多数pn是良性的,但一些是恶性的,导致另外的干预。对于视为关于恶性结节的低风险的患者,当前的医学实践要求每三至六个月的扫描,共至少两年,以监测肺癌。pn鉴定至诊断之间的时间段是医学监视或“观察等待”的时间,并且可能造成对患者的压力,并且由于重复的成像研究,导致显著的风险和费用。如果对发现具有良性结节的患者执行活组织检查,则不必要地增加了成本和伤害患者的可能性。需要大手术,以便切除样本用于组织活组织检查和诊断。所有这些程序都与患者的风险相关,所述风险包括:病、损伤和死亡、以及高经济成本。通常,由于其大小和/或在肺中的位置,pn无法进行活组织检查,以确定它们是良性的还是恶性的。然而,pn连接至循环系统,并且因此如果是恶性的,则癌症的蛋白质标记物可以进入血液,并且提供信号用于确定pn是否为恶性的。需要可以替换补充用于呈现有pn的患者的当前诊断方法的诊断方法,以改善诊断、降低成本并且使对患者的侵入性程序和并发症降到最低。本发明提供了用于鉴定蛋白质标记物的新型组合物、方法和试剂盒,以鉴定、诊断、分类和监测肺部状况,且特别是肺癌。本发明使用基于血液的复用测定来区分良性肺结节与恶性肺结节,以分类患有或不患有肺癌的患者。本发明可以用于呈现有肺癌症状,但没有肺结节的患者。概述本发明提供了通过以下确定受试者中的肺部状况为癌症的可能性的方法:测量从受试者获得的样品中的蛋白质实验对象组的丰度;基于蛋白质测量计算癌症评分的概率,并且如果该评分低于预定评分,则对于受试者排除癌症。当排除癌症时,受试者不接受治疗方案。治疗方案包括例如肺功能测试(pft),肺成像,活组织检查,手术,化学疗法,放射疗法或其任何组合。在一些实施方案中,成像是x射线、胸部计算机断层摄影(ct)扫描、或正电子发射断层摄影(pet)扫描。本发明进一步提供了通过以下判定受试者的癌症可能性的方法:测量从受试者获得的样品中的蛋白质实验对象组的丰度,基于蛋白质测量计算癌症评分的概率,并且如果该评分高于预定评分,则对于受试者判定癌症的可能性。在另一个方面,本发明进一步提供了通过以下确定受试者中存在肺部状况的可能性的方法:测量从受试者获得的样品中的蛋白质实验对象组的丰度,基于蛋白质测量计算癌症评分的概率,并且如果该评分等于或大于预定评分,则得出结论存在所述肺部状况。肺部状况是肺癌,例如非小细胞肺癌(nsclc)。受试者处于发展肺癌的风险中。在另一个方面,本发明提供了确定受试者中的肺结节不是肺癌的可能性的方法,其包括:(a)测量从受试者获得的血液样品中存在的蛋白质实验对象组的表达水平,其中所述蛋白质实验对象组包含lg3bp和c163a、基本上由lg3bp和c163a组成、或由lg3bp和c163a组成;(b)基于步骤(a)的蛋白质实验对象组的表达水平,计算肺癌评分的概率;并且(c)如果步骤(b)中的评分低于预定评分,则对于受试者排除肺癌。在一些实施方案中,实验对象组包括选自以下的至少3种蛋白质:aldoa、fril、lg3bp、ibp3、lrp1、islr、tsp1、coia1、grp78、tetn、prdx1和cd14。任选地,实验对象组进一步包括选自bgh3、coia1、tetn、grp78、prdx、fiba和gslg1的至少一种蛋白质。在一些实施方案中,实验对象组包括选自以下的至少4种蛋白质:aldoa、fril、lg3bp、ibp3、lrp1、islr、tsp1、coia1、grp78、tetn、prdx1和cd14。在一个优选实施方案中,实验对象组包含lrp1、coia1、aldoa和lg3bp。在另一个优选实施方案中,实验对象组包含lrp1、coia1、aldoa、lg3bp、bgh3、prdx1、tetn和islr。在又一个优选实施方案中,实验对象组包含lrp1、coia1、aldoa、lg3bp、bgh3、prdx1、tetn、islr、tsp1、grp78、fril、fiba和gslg1。受试者具有或怀疑具有肺结节。肺结节具有小于或等于3cm的直径。在一个实施方案中,肺结节具有约0.8cm至2.0cm的直径。从应用于蛋白质测量的逻辑回归模型计算评分。例如,评分确定为,其中是所述样品(s)中对数转换并标准化的跃迁强度,是相应的对数回归系数,是实验对象组特异性常数,并且n是所述实验对象组中的跃迁总数目。在各个实施方案中,本发明的方法进一步包括将蛋白质测量标准化。例如,蛋白质测量通过选自pedf、masp1、gels、lum、c163a和ptprj的一种或多种蛋白质进行标准化。生物样品包括例如组织、血液、血浆、血清、全血、尿、唾液、生殖器分泌物、脑脊髓液、汗和排泄物。在一个方面,确定癌症的可能性通过与评分相关的灵敏度、特异性、阴性预测值或阳性预测值来确定。确定的评分具有至少约80%的阴性预测值(npv)。测量步骤通过使用选择性反应监测质谱法来执行,其使用特异性结合待检测蛋白质的化合物或肽跃迁。在一个实施方案中,与待测量蛋白质特异性结合的化合物是抗体或适体。附图简述图1是显示了关于15个蛋白质lc-srm-ms实验对象组的接受操作曲线的曲线下面积的线图。图2显示了六个线图,其各自显示了对于不同的患者群体以及具有大pn和小pn的受试者,关于15个蛋白质lc-srm-ms实验对象组的接受操作曲线的曲线下面积。图3是显示了用于评估15个蛋白质实验对象组的三项研究中的变化性的图。图4是显示了关于15个蛋白质lc-srm-ms实验对象组的接受操作曲线的曲线下面积的线图。图5显示了三个线图,其各自显示了对于不同的患者群体,关于15个蛋白质lc-srm-ms实验对象组的接受操作曲线的曲线下面积。图6显示了使用"ingenuity"®程序,用于鉴定肺癌的血液蛋白质的查询结果。图7是显示了关于来自相同肽、相同蛋白质和不同蛋白质的肽的皮尔森相关性的条形图。图8是显示了分类器对训练样品、验证样品和组合的所有样品的性能的图。图9是显示了临床因素和分子因素的图。图10是示意图,其显示了含有肺癌、对氧化性应激的应答和肺部炎症的13种分类器蛋白(绿色)、5种转录因子(蓝色)和3种网络(橙色线)的分子网络。图11是描绘了根据风险来解释分类器评分的图。图12是显示了分类器对发现样品(n=143)和验证样品(n=104)的性能的图。阴性预测值(npv)和特异性(spc)根据分类器评分呈现。假设20%的癌症流行率。图13是显示了临床因素(吸烟、结节大小)和分子因素(分类器评分)的多变量分析的图,因为它们在发现和验证研究中与癌症和良性样品(n=247)相关。吸烟在垂直线上按包年进行测量。结节大小由圆直径表示。呈现了0.43的参考值,以说明与高于参考值的大量癌症样品相比,在低于参考值的少量癌症样品之间的区别。图14是显示了肺癌、氧化性应激应答和肺部炎症的13种分类器蛋白(绿色)、4种转录调节剂(蓝色)和3种网络(橙色线)的图。所有参考都是人uniprot标识符。图15是显示了所有247个患者的结节大小相对于分类器评分的散布图的图,其证实了两个变量之间的相关性缺乏。图16是显示了关于来自相同肽(蓝色)、相同蛋白质(绿色)和不同蛋白质(红色)的肽的皮尔逊相关性的图解。图17是显示了log2elisa浓度比(半乳凝素3bp/cd163a)相对于质谱法比率(半乳凝素3bp/cd163a)的log2的相关性的图。图18是显示了xl1w校准的历史分布的图。图19是显示了xl2逆转评分历史分布的图。详述本发明衍生自以下令人惊讶的发现:在呈现有肺结节的患者中,血液中存在特异性鉴定且分类肺癌的蛋白质标记物。相应地,本发明对患者提供了与患者中的肺癌早期检测相关的独特优点,包括寿命增加、发病率和死亡率降低、在筛查和重复筛查期间减少的辐射暴露以及微创诊断模型。重要的是,本发明的方法允许患者避免侵入性程序。胸部计算机断层摄影(ct)扫描的常规临床应用每年鉴定了数百万个肺结节,其中仅少数是恶性的,但对于诊断为非小细胞肺癌(nsclc)的患者,促成惨淡的15%五年存活率。具有肺结节的患者中的肺癌早期诊断是当务之急,因为与当前的非侵入性诊断选项(例如胸部ct和正电子发射断层摄影(pet)扫描)以及其它侵入性替代方案结合,基于临床表现的决策制定并未改变i期nsclc患者的临床结果。相对于8mm以下的恶性肿瘤的较低发生率和20mm以上的恶性肿瘤的较高发生率,大小在8mm至20mm之间的肺结节亚组越来越多地被识别为“中等的”[9]。通过使用经胸壁针刺抽吸的活组织检查或支气管镜检查的肺部结节的侵入性取样,可以提供nsclc的细胞病理学诊断,但也与假阴性和非诊断性结果两者相关。总之,对于肺结节管理的关键的未满足的临床需要是非侵入性的诊断测试,其区别具有不确定肺结节(ipn)的患者中的恶性和良性过程,所述不确定肺结节尤其是大小在8mm至20mm之间。在治疗上或多或少攻击性的临床决策基于危险因素,主要是结节大小、吸烟史和年龄[9]加上成像。由于这些不是结论性的,因此非常需要基于分子的血液测试,其既是非侵入性的,又对危险因素和成像提供补充信息。相应地,这些和相关实施方案在用于肺部状况,且特别是肺癌诊断的筛查方法中有用。更重要的是,本发明在确定患者的临床管理中有用。即,本发明的方法可用于对于各个受试者判定或排除特定的治疗方案。癌症生物学需要分子策略来解决用于肺癌风险评价的未满足的医学需要。诊断医学领域已随着技术和测定而进化,所述技术和测定提供了用于检测蛋白质中的变化的灵敏机制。本文所述的方法使用lc-srm-ms技术,用于测量在具有恶性pn的患者中共同改变的血浆蛋白的浓度。这种蛋白质标记指示肺癌。lc-srm-ms是提供血浆中的循环蛋白质的定量和鉴定两者的一种方法。蛋白质表达水平中的变化,例如但不限于信号转导因子、生长因子、切割的表面蛋白和分泌性蛋白,可以使用测定癌症的此类灵敏技术来检测。本文呈现的是基于血液的分类测试,以确定呈现有肺结节的患者具有良性或恶性结节的可能性。本发明呈现了预测了pn是良性还是恶性的相对可能性的分类算法。更广泛地,证实了存在关于本发明的许多变化,其也是关于pn是良性还是恶性的可能性的诊断测试。这些是关于蛋白质实验对象组、蛋白质标准、测量方法学和/或分类算法的变化。如本文公开的,通过质谱法就差异蛋白质表达分析来自呈现有pn的受试者的存档血浆样品,并且结果用于鉴定生物标记物蛋白质和生物标记物蛋白质的实验对象组,其与各种肺部状况(癌症相对于非癌症)结合是差异表达的。在本发明的一个方面,发现了163个实验对象组,其允许将pn分类为良性或恶性的。这些实验对象组包括表1上列出的实验对象组。在一些实施方案中,根据本发明的实验对象组包括测量选自以下的1、2、3、4、5种或更多种蛋白质:islr、aldoa、kit、grp78、aifm1、cd14、coia1、ibp3、tsp1、bgh3、tetn、fri、lg3bp、ggh、prdx1或lrp1。在其它实施方案中,实验对象组包括表1上例示的任何实验对象组或蛋白质。例如,实验对象组包括aldoa、grp78、cd14、coia1、ibp3、fril、lg3bp和lrp1。用于判定受试者的治疗的优选实验对象组包括表3和4上列出的实验对象组。在各种其它实施方案中,根据本发明的实验对象组包括测量表2和3上列出的至少2、3、4、5、6、7种或更多种蛋白质。表3表4表5中列出了优选的标准化物(normalizer)实验对象组。表5术语“肺结节”(pn)指可以通过射线照相技术显现的肺部病变。肺结节是直径小于或等于三厘米的任何结节。在一个实例中,肺结节具有约0.8cm至2cm的直径。术语“肿块”或“肺肿块”指最大直径大于三厘米的肺部结节。术语“血液活组织检查”指血液的诊断研究,以确定呈现有结节的患者是否具有可以分类为良性或恶性的状况。术语“接受标准”指这样的一组标准,测定、测试、诊断或产品应该符合所述标准,以对于其预期用途视为可接受的。如本文使用的,接受标准是测试、对分析程序的提及以及适当测量的列表,其对于用于诊断中的测定或产品进行限定。例如,关于分类器的接受标准指一组预定范围的系数。术语“平均最大auc”指计算性能的方法学。对于本发明,在通过向前或向后选择限定应该在实验对象组中的蛋白质集合的过程中,一次去除或添加一种蛋白质。可以生成关于性能(y轴上的auc或部分auc评分和x轴上的蛋白质)的图,使性能达到最大的点指示给出最佳结果的蛋白质数目和集合。术语“部分auc因子或pauc因子”大于通过随机预测所预期的。在灵敏度=0.90下,pauc因子是从0.9到1.0特异性/(0.1*0.1/2)的梯形roc曲线下面积。术语“增量信息”指可以与其它诊断信息一起使用,以增强诊断准确度的信息。增量信息不依赖于临床因素,例如包括结节大小、年龄或性别。术语“评分(score)”或“评分(scoring)”指计算关于样品的概率可能性。对于本发明,接近于1.0的值用于表示样品是癌症的可能性,接近于0.0的值表示样品是良性的可能性。术语“稳固的”指不因违反它基于其的假设而严重扰乱的测试或程序。对于本发明,稳固的测试是这样的测试,其中已手动审查了质谱法层析图的蛋白质或跃迁,并且“一般”不含干扰信号。术语“系数”指分配给逻辑回归方程中用于对样品评分的每种蛋白质的权重。在本发明的某些实施方案中,考虑到根据mccv的逻辑回归模型,取决于蛋白质分类器的测量方法(或模型),模型系数和每种蛋白质的模型系数的变异系数(cv)可以增加或减少。对于实验对象组中列出的每种蛋白质,对于系数和cv各自存在约、至少、至少约或至多约2、3、4、5、6、7、8、9或10倍或其中可衍生的任何范围。可替代地,考虑本发明的定量实施方案可以根据约、至少、至少约或至多约10、20、30、40、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99%或更多、或其中可衍生的任何范围进行讨论。术语“最佳团队合作者”指在随机实验对象组选择算法中排名最高的蛋白质,即,对实验对象组表现良好的蛋白质。当组合到分类器内时,这些蛋白质可以将癌症与良性样品分离。“最佳团队合作者”蛋白与“协同蛋白”同义。术语“协同蛋白”指在蛋白质的高性能实验对象组上,比偶然预期的更频繁地出现的蛋白质。这产生蛋白质的协同评分,其测量蛋白质在高性能实验对象组上多么(不)频繁地出现。例如,协同评分为1.5的蛋白质在高性能实验对象组上出现的概率是单独偶然预期的1.5x。如本文使用的,关于肺部状况的术语“分类”指使用统计技术汇编且分析表达数据的行为,以提供分类来帮助诊断肺部状况,特别是肺癌。如本文使用的,术语“分类器”指以预定水平的统计显著性区别疾病状态的算法,两类分类器是使用来自样品的测量的数据点,并且将数据分类为两组之一的算法。在某些实施方案中,分类器中使用的数据是生物样品中蛋白质的相对表达,受试者中的蛋白质表达水平可以与先前诊断为无疾病或指定状况的患者中的水平进行比较。“分类器”使区分随机选择的癌症样品与随机选择的良性样品的概率达到最大,即roc曲线的auc。除具有差异表达的分类器的组成成分蛋白质之外,它还可以包括具有最低限度的生物学变异或没有生物学变异的蛋白质,以允许评价临床样本内或临床样本之间的变异性或变异性的缺乏;这些蛋白质可以称为内源蛋白质,并且充当其它分类器蛋白质的内部对照。如本文使用的,术语“标准化”或“标准化物”指根据标准值的差异值表述,以调整由于样品处理、样品制备和质谱法测量而起于技术变化,而不是样品中的蛋白质浓度的生物学变异的效应。例如,当测量差异表达的蛋白质的表达时,关于该蛋白质的表达的绝对值可以根据关于标准蛋白质的表达的绝对值来表达,所述标准蛋白质在表达方面是基本上恒定的。这防止样品制备和质谱法测量的技术变化妨碍样品中的蛋白质浓度水平的测量。如本文使用的,术语“状况”一般指疾病、事件或健康状况中的改变。如本文使用的,术语“治疗方案”包括通常执行的进一步的诊断测试,以确定肺结节是良性还是恶性的。治疗方案包括通常用于诊断肺结节或肿块的诊断测试,例如ct扫描、正电子发射断层摄影(pet)扫描、支气管镜检查或组织活组织检查。如本文使用的,治疗方案还意欲包括通常用于治疗恶性肺结节和/或肺癌的治疗性治疗,例如化学疗法、放射疗法或外科手术。术语“诊断”和“诊断学”还分别涵盖术语“预后(prognosis)”和“预后(prognostics)”,以及此类程序在两个或更多个时间点上的应用,以随着时间过去监测诊断和/或预后,并且基于此进行统计建模。此外,术语诊断包括:a.预测(确定患者是否可能发展过度增殖性疾病),b.预后(预测患者在将来的预先选择的时间可能具有更好还是更坏的结果),c.疗法选择,d.治疗药物监测,复发监测。在一些实施方案中,例如,将生物样品分类为衍生自患有肺部状况的受试者可以指由实验室生成的结果和相关报告,而诊断可以指医学专业人员使用分类法来鉴定或验证肺部状况的行为。如本文使用的,关于生物样品的术语“提供”指从受试者直接或间接地获得生物样品。例如,“提供”可以指从受试者直接获得生物样品的行为(例如,通过抽血、组织活组织检查、灌洗等等)。同样地,“提供”可以指间接地获得生物样品的行为。例如,提供可以指实验室从直接获得样品的一方接受样品的行为,或者从存档获得样品的行为。如本文使用的,“肺癌”优选指肺部的癌症,但可以包括人或其它哺乳动物的呼吸系统的任何疾病或其它病症。呼吸系统赘生性病症包括例如小细胞癌或小细胞肺癌(sclc)、非小细胞癌或非小细胞肺癌(nsclc)、鳞状细胞癌、腺癌、支气管肺泡癌、混合性肺癌、恶性胸膜间皮瘤、未分化大细胞癌、巨细胞癌、同时性肿瘤、大细胞神经内分泌癌、腺鳞状癌、未分化癌;以及小细胞癌,包括燕麦细胞癌、混合性小细胞/大细胞癌和复合型小细胞癌;以及腺样囊性癌、错构瘤、粘液表皮样肿瘤、典型类癌性肺肿瘤、非典型类癌性肺肿瘤、周围类癌性肺肿瘤、中心类癌性肺肿瘤、胸膜间皮瘤和未分化肺癌以及源自肺外的癌症,例如已从身体的其它部分转移到肺部的继发性癌症。肺癌可以是任何阶段或等级。优选地,该术语可以用于共同地指任何异型增生、增生、赘瘤形成或转移,其中可以例如通过与邻近的健康组织的比较,来确定表达高于正常水平的蛋白质生物标记物。非癌性肺部状况的实例包括慢性阻塞性肺疾病(copd)、良性肿瘤或细胞肿块(例如错构瘤、纤维瘤、神经纤维瘤)、肉芽肿、结节病、以及由细菌(例如结核病)或真菌(例如组织胞浆菌病)病原体引起的感染。在某些实施方案中,肺部状况可能与射线照相pn的出现有关。如本文使用的,“肺组织”和“肺癌”分别指肺本身以及与肺下面的层相邻和/或在其内的组织和支撑结构(例如胸膜、肋间肌、肋骨和呼吸系统的其它元件)的组织或癌症。在此上下文中,呼吸系统本身视为代表鼻腔、窦、咽、喉、气管、支气管、肺、肺叶、肺泡、肺泡管、肺泡囊、肺泡毛细血管、细支气管、呼吸性细支气管、脏层胸膜、壁层胸膜、胸膜腔、横膈膜、会厌、增殖腺、扁桃体、口和舌等等。组织或癌症可以来自哺乳动物,且优选来自人,尽管猴、猿、猫、犬、牛、马和兔在本发明的范围内。如本文使用的,术语“肺部状况”指与肺相关的疾病、事件或健康状况中的改变,包括例如肺癌和各种非癌性状况。“准确度”指测量或计算的数量(测试报告值)与其实际(或真实)值的符合程度。临床准确度涉及真实结果(真阳性(tp)或真阴性(tn)相对于错误分类的结果(假阳性(fp)或假阴性(fn))的比例,并且可以陈述为灵敏度、特异性、阳性预测值(ppv)或阴性预测值(npv),或者可能性、比值比以及其它测量结果。如本文使用的,术语“生物样品”指潜在地含有一种或多种生物标记物蛋白质的生物起源的任何样品。生物样品的实例包括组织,器官或体液,例如全血、血浆、血清、组织、灌洗液或用于疾病检测的任何其它样本。如本文使用的,术语“受试者”指哺乳动物,优选人。如本文使用的,术语“生物标记物蛋白质”指来自患有肺部状况的受试者的生物样品相对于来自对照受试者的生物样品中的多肽。生物标记物蛋白质不仅包括多肽本身,还包括其较小的变异,包括例如一个或多个氨基酸取代或修饰,例如糖基化或磷酸化。如本文使用的,术语“生物标记物蛋白质实验对象组”指多种生物标记物蛋白质。在某些实施方案中,实验对象组中蛋白质的表达水平可以与受试者中肺部状况的存在相关联。在某些实施方案中,生物标记物蛋白质实验对象组包含2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、60、70、80、90或100种蛋白质。在某些实施方案中,生物标记物蛋白质实验对象组包含100-125种蛋白质、125-150种蛋白质、150-200种蛋白质或更多。如本文使用的,关于状况的“治疗(treating)”或“治疗(treatment)”可以指预防状况,减缓状况的发作或发展速率,降低发展状况的风险,预防或延迟与状况有关的症状的发展,降低或终止与状况有关的症状,生成状况的完全或部分消退或其某种组合。如本文使用的,术语“排除”意指选择不接受治疗方案的受试者。如本文使用的,术语“判定”意指选择接受治疗方案的受试者。生物标记物水平可以由于疾病的治疗而改变。生物标记物水平中的改变可以通过本发明进行测量。生物标记物水平中的改变可以用于监测疾病或疗法的进展。“变更的”、“改变的”或“显著不同的”指与合理地可比较状态、概况、测量等等的可检测改变或差异。本领域技术人员应该能够确定合理的可测量改变。此类改变可以是全或无。它们可以是增量的,而无需是线性的。它们可能是数量级的。改变可以是1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%、99%、100%或更多、或0%至100%之间的任何值的增加或减少。可替代地,改变可以是1倍、1.5倍、2倍、3倍、4倍、5倍或更多倍,或者1倍至5倍之间的任何值。改变可能是统计学显著的,具有0.1、0.05、0.001或0.0001的p值。使用本发明的方法,首先执行患者的临床评价。如果存在更高的癌症可能性,则临床医生可以判定要求寻找诊断测试选项的疾病,产生增加和/或确证诊断可能性的数据。疾病的“判定”要求具有高特异性的测试。“fn”是假阴性,对于疾病状态测试,其意指将疾病受试者错误地分类为非疾病或正常。“fp”是假阳性,对于疾病状态测试,其意指将正常受试者错误地分类为患有疾病。术语“判定”指具有高特异性的诊断测试,其加上临床评价指示更高的癌症可能性。如果临床评价是更低的癌症可能性,则临床医生可以采取排除疾病的立场,其要求产生减少诊断可能性的数据的诊断测试。“排除”要求具有高灵敏度的测试。术语“排除”指具有高灵敏度的诊断测试,其加上临床评价指示更低的癌症可能性。术语“测试的灵敏度”指患有疾病的患者具有阳性测试结果的概率。这衍生自具有阳性测试结果(真阳性)的患有疾病的患者数目除以患有疾病的患者总数目,包括具有真阳性结果的患者和具有阴性结果(即假阴性)的患有疾病的患者总数目。术语“测试的特异性”指没有疾病的患者具有阴性测试结果的概率。这衍生自具有阴性测试结果(真阴性)的没有疾病的患者数目除以没有疾病的所有患者,包括具有真阴性结果的患者和具有阳性测试结果(例如假阳性)的没有疾病的患者。虽然测试的灵敏度、特异性、真阳性率或假阳性率、以及真阴性率或假阴性率,提供了指示测试性能(例如相对于其它测试)的指示,以基于测试的结果对于各个患者做出临床决策,临床医生要求关于给定群体的测试性能参数。术语“阳性预测值”(ppv)指阳性结果正确地鉴定患有疾病的患者的概率,其是真阳性的数目除以真阳性和假阳性的总和。术语“阴性预测值”或"npv"通过tn/(tn+fn)或所有阴性测试结果的真阴性分数进行计算。它也固有地受到疾病的流行率和预期测试的群体的测试前概率的影响。术语“疾病流行率”指在特定时期过程中疾病的所有新老病例或事件发生的数目。流行率表示为其中事件数目是分子且处于风险中的群体是分母的比率。术语疾病发生率指在指定时间段内发展某种新状况的风险的量度;在某一时间段期间的新病例数目,它更好地表示为与分母的比例或比率。根据“美国国家肺癌筛查测试(nationallungscreeningtrial)”的肺癌风险按年龄和吸烟史分类。高风险-年龄≥55和≥30包年的吸烟史;中等风险–年龄≥50和≥20包年的吸烟史;低风险-<50岁或<20包年的吸烟史。术语“阴性预测值”(npv)指阴性测试正确地鉴定没有疾病的患者的概率,其是真阴性的数目除以真阴性和假阴性的总和。如果关于患者可以视为其部分的给定群体的疾病流行率是已知的,则来自具有足够ppv的测试的阳性结果可以用于判定患者的疾病,而来自具有足够npv的测试的阴性结果可以用于排除疾病。临床医生必须使用诊断测试来作出决定,所述诊断测试基于其固有性能参数包括灵敏度和特异性、以及其外部性能参数例如阳性预测值和阴性预测值,所述参数取决于疾病在给定群体中的流行率。可能影响疾病可能性的临床评价的另外参数包括患者与已知因子(例如暴露风险)的先前频率和亲密性,所述因子与疾病原因直接或间接地有关,例如二手烟、辐射等,以及肺结节的射线照相表现或特征(不计大小)。结节的描述可以包括固体、半固体或磨玻璃,其基于由ct扫描技术所采用的相对灰阶密度谱来表征。“质谱法”指这样的方法,其包括采用电离源,由探针的样品呈递表面上呈递的分析物生成气相离子,并且用质谱仪检测气相离子。技术液相层析选择性反应监测质谱法(lc-srm-ms)用于测定血液中的388种蛋白质组群的表达水平,以鉴定可能与疾病的不存在或存在相关联的个别蛋白质的差异。个别蛋白质不仅已牵涉肺癌生物学,而且还可能基于其作为膜锚定或分泌蛋白的表达而存在于血浆中。切除的肺癌组织(包括腺癌、鳞状和大细胞亚型)的上皮和内皮膜的分析鉴定了217种组织蛋白。用与肺癌生物学相关的关键词的科学文献审查鉴定了319种蛋白质。通过癌组织分析或文献审查鉴定的蛋白质之间存在148种蛋白质的重叠,产生了总共388种独特蛋白质作为候选物。在来自新鲜nsclc切除和邻近非恶性组织的分泌囊内容物的蛋白质组学分析之后,发现了包括在多路lc-srm-ms测定中的大多数候选蛋白质。使用广泛的生物信息学和文献注释,鉴定了在肿瘤组织中可再现地上调的分泌性蛋白,并且区分优先次序用于包括在lc-srm-ms测定中。相关文献中存在的另外一组蛋白质也加入测定中。总共,与肺癌相关的388种蛋白质被区分优先次序用于srm测定开发。在这些中,371种候选蛋白质生物标记物最终包括在测定中。这些在下表6中列出。表6.这些候选蛋白质生物标记物中的190种显示为在血液中可再现地测量。来自pn患者的242个血液样品的适度操纵的多场所无偏研究,设计为确定是否可以鉴定统计学显著的蛋白质子实验对象组,以区分大小在2cm以下的良性和恶性结节。对该研究贡献样品和临床数据的三个场所是universityoflaval、universityofpennsylvania和newyorkuniversity。在本发明的一个实施方案中,15种蛋白质的实验对象组有效地区分衍生自具有直径小于2cm的良性和恶性结节的患者的样品。生物信息学和生物统计分析首先用于鉴定具有统计学显著差异表达的个别蛋白质,然后使用这些蛋白质来衍生一种或多种蛋白质组合或蛋白质实验对象组,与任何个别蛋白质相比,其共同地证实优异的区别性能。生物信息学和生物统计方法用于衍生关于实验对象组中的每种个别蛋白质的系数(c),这反映其相对表达水平即增加或减少,以及相对于其它蛋白质,在实验对象组的净区别能力方面的权重或重要性。实验对象组的定量区别能力可以表示为数学算法,其中关于其组成成分蛋白各自的项是其系数与蛋白质血浆表达水平(p)(如通过lc-srm-ms测量的)的乘积,例如cxp,其中由n种蛋白质组成的算法描述为:c1xp1+c2xp2+c3xp3+…+cnxpn。以预定水平的统计显著性区别疾病状态的算法可以被称为“疾病分类器”。除具有差异表达的分类器的组成成分蛋白质之外,它还可以包括具有最低限度的生物学变异或没有生物学变异的蛋白质,以允许评价临床样本内或临床样本之间的变异性或变异性的缺乏;这些蛋白质可以称为典型的天然蛋白质,并且充当其它分类器蛋白质的内部对照。在某些实施方案中,表达水平通过ms进行测量。ms基于其质荷比,分析在其通过其亲本蛋白质的汽化而产生及其与其它离子分开后,由离子所产生的质谱。采集ms数据的最常见模式是:1)全扫描采集,导致典型的总离子流图(tic),2)选择性离子监测(sim),以及3)选择性反应监测(srm)。在本文提供的方法的某些实施方案中,生物标记物蛋白质表达水平通过lc-srm-ms进行测量。lc-srm-ms是串联质谱的高度选择性方法,其具有有效地过滤出除了所需分析物之外的所有分子和污染物的潜力。如果分析样品是复杂的混合物,其在限定的分析窗口内可能包含几种同质量(isobaric)种类,则这是特别有益的。lc-srm-ms方法可以利用三重四极杆质谱仪,如本领域中已知的,所述质谱仪包括三个四极杆集。在第一四极杆集中执行质量选择的第一阶段,并且在第二四极杆集中将选择性传输的离子片段化。所得的跃迁(产物)离子被输送到第三四极杆集,其执行质量选择的第二阶段。通过检测器测量通过第三四极杆集传输的产物离子,所述检测器生成代表选择性传输的产物离子数目的信号。调谐施加到第一四极杆和第三四极杆的rf和dc电位,以(分别)选择具有位于指定的窄范围内的m/z值的前体离子和产物离子。通过指定适当的跃迁(前体离子和产物离子的m/z值),可以以高度灵敏度和选择性测量对应于靶向蛋白质的肽。信噪比优于传统的串联质谱法(ms/ms)实验,其在第一个四极杆中选择一个质量窗口,然后测量离子检测器中所有生成的跃迁。lc-srm-ms。在某些实施方案中,如本文公开的用于诊断或监测肺癌的srm-ms测定,可以利用衍生自表6所示蛋白质的一种或多种肽和/或肽跃迁。在某些实施方案中,该测定可以利用来自100种或更多种、150种或更多种、200种或更多种、250种或更多种、300种或更多种、345种或更多种、或者371种或更多种生物标记物蛋白质的肽和/或肽跃迁。在某些实施方案中,每种生物标记物蛋白质可以利用两种或更多种肽,并且在这些实施方案的某些中,可以利用四种或更多种肽中的三种或更多种。类似地,在某些实施方案中,每种肽可以利用两个或更多个跃迁,并且在这些实施方案的某些中,每种肽可以利用三个或更多个;四个或更多个;或者五个或更多个跃迁。在一个实施方案中,用于诊断肺癌的lc-srm-ms测定可以测量五个跃迁的强度,所述五个跃迁对应于与每种生物标记物蛋白质有关的选定肽。可以根据在这种分析过程中观察到的信号强度,对于每种肽估计可达到的定量极限(loq)。例如,关于与肺癌有关的靶蛋白集合,参见表12。可以使用本领域已知的任何合适方法来测量生物标记物蛋白质的表达水平,所述方法包括但不限于质谱法(ms)、逆转录酶-聚合酶链反应(rt-pcr)、微阵列、基因表达系列分析(sage)、通过大规模平行标记测序(mpss)的基因表达分析、免疫测定(例如elisa)、免疫组织化学(ihc)、转录组学和蛋白质组学。当elisa用于测量生物标记物蛋白质的表达水平时,可以使用特异性结合生物标记物蛋白质的抗体。例如,lg3bp抗体用于测量lg3bp的表达水平;c163a抗体用于测量c163a的表达水平。在一些实施方案中,该方法包括使得自受试者的血液样品与lg3bp抗体和c163a抗体接触。为了评估肽跃迁的特定集合的诊断性能,对于每个显著跃迁生成roc曲线。如本文使用的,“roc曲线”指当二元分类器系统的区别阈值变化时,关于其的真阳性率(灵敏度)针对假阳性率(特异性)的图。roc曲线可以通过绘制阳性中的真阳性分数(tpr=真阳性率)相对于阴性中的假阳性分数(fpr=假阳性率)来等价地表示。roc曲线上的每个点代表对应于特定决策阈值的灵敏度/特异性对。图7和9提供了在患病受试者的组群和非患病受试者的组群中的生物标记物或生物标记物实验对象组灵敏度和特异性值的分布之间的功能关系的图形表示。auc代表roc曲线下面积。auc是1)生物标记物或生物标记物实验对象组和2)roc曲线的诊断准确度的总体指标。auc由“梯形法则”确定。对于给定的曲线,数据点通过直线段连接,垂直线从横坐标竖立到每个数据点,并且计算如此构建的三角形和梯形面积的总和。在本文提供的方法的某些实施方案中,生物标记物蛋白质具有在约0.75至1.0的范围内的auc。在这些实施例的某些中,auc在约0.8至0.8、0.9至0.95、或0.95至1.0的范围内。本文提供的方法是微创性的并且造成很少的不利效应或没有不利效应的风险。像这样,它们可以用于诊断、监测且提供并未显示出肺部状况的任何症状的受试者、以及被分类为对于发展肺部状况的低风险的受试者的临床管理。例如,本文公开的方法可以用于诊断受试者中的肺癌,所述受试者并未呈现有pn和/或过去并未呈现有pn,但仍然被认为处于发展pn和/或肺部状况的风险中。类似地,本文公开的方法可以用作严格的预防措施,以诊断被分类为对于发展肺部状况的低风险的健康受试者。本发明提供了通过以下确定受试者中的肺部状况为癌症的可能性的方法:测量从受试者获得的样品中的蛋白质实验对象组的丰度;基于蛋白质测量计算癌症评分的概率,并且如果该评分低于预定评分,则对于受试者排除癌症,其中当排除癌症时,受试者不接受治疗方案。治疗方案包括例如肺功能测试(pft),肺成像,活组织检查,手术,化学疗法,放射疗法或其任何组合。在一些实施方案中,成像是x射线、胸部计算机断层摄影(ct)扫描、或正电子发射断层摄影(pet)扫描。本发明进一步提供了通过以下判定受试者的癌症可能性的方法:测量从受试者获得的样品中的蛋白质实验对象组的丰度,基于蛋白质测量计算癌症评分的概率,并且如果步骤中的该评分高于预定评分,则对于受试者判定癌症的可能性。在另一个方面,本发明进一步提供了通过以下确定受试者中存在肺部状况的可能性的方法:测量从受试者获得的样品中的蛋白质实验对象组的丰度,基于蛋白质测量计算癌症评分的概率,并且如果该评分等于或大于预定评分,则得出结论存在所述肺部状况。肺部状况是肺癌,例如非小细胞肺癌(nsclc)。实验对象组包括选自aldoa、fril、lg3bp、ibp3、lrp1、islr、tsp1、coia1、grp78、tetn、prdx1和cd14的至少4种蛋白质。任选地,实验对象组进一步包括选自bgh3、coia1、tetn、grp78、prdx、fiba和gslg1的至少一种蛋白质。可替代地,实验对象组包括选自aldoa、fril、lg3bp、ibp3、lrp1、islr、tsp1、coia1、grp78、tetn、prdx1和cd14的至少3种蛋白质。在一些实施方案中,实验对象组包含选自lrp1、coia1、aldoa和lg3bp的至少1、2、3或4种蛋白质。在一些实施方案中,实验对象组包含选自lrp1、coia1、aldoa、lg3bp、bgh3、prdx1、tetn和islr的至少1、2、3、4、5、6、7或8种蛋白质。在一些实施方案中,实验对象组包含选自lrp1、coia1、aldoa、lg3bp、bgh3、prdx1、tetn、islr、tsp1、grp78、fril、fiba、gslg1的至少1、2、3、4、5、6、7、8、9、10、11、12或13种蛋白质。任选地,实验对象组包括选自tsp1、coia1、islr、tetn、fril、grp78、aldoa、bgh3、lg3bp、lrp1、fiba、prdx1、gslg1、kit、cd14、ef1a1、tenx、aifm1、ggh、ibp3、enpl、ero1a、6pgd、icam1、ptpa、ncf4、sem3g、1433t、rap2b、mmp9、folh1、gstp1、ef2、ran、sodm和dsg2的至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35或36种蛋白质。任选地,实验对象组包括选自fril、tsp1、lrp1、prdx1、tetn、tbb3、coia1、ggh、a1ag1、aifm1、ampn、crp、gslg1、ibp3、kit、nrp1、6pgd、ch10、clic1、cof1、csf1、cytb、dmkn、dsg2、ereg、ero1a、folh1、ileu、k1c19、lyox、mmp7、ncf4、pdia3、ptgis、ptpa、ran、scf、sem3g、tba1b、tcpa、tera、timp1、tnf12和ugpa的至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44种蛋白质。受试者具有或怀疑具有肺结节。肺结节具有小于或等于3cm的直径。在一个实施方案中,肺结节具有约0.8cm至2.0cm的直径。受试者可能患有ia期肺癌(即,肿瘤小于3cm)。从应用于蛋白质测量的逻辑回归模型计算评分。例如,评分确定为其中是所述样品(s)中对数转换并标准化的跃迁强度,是相应的对数回归系数,是实验对象组特异性常数,并且n是所述实验对象组中的跃迁总数目。在各个实施方案中,本发明的方法进一步包括将蛋白质测量标准化。例如,蛋白质测量通过选自pedf、masp1、gels、lum、c163a和ptprj的一种或多种蛋白质进行标准化。生物样品例如组织、血液、血浆、血清、全血、尿、唾液、生殖器分泌物、脑脊髓液、汗和排泄物。在一个方面,确定癌症的可能性通过与评分相关的灵敏度、特异性、阴性预测值或阳性预测值来确定。确定的评分具有至少约60%、至少70%或至少约80%的阴性预测值(npv)。测量步骤通过使用选择性反应监测质谱法来执行,所述方法使用特异性结合待检测蛋白质的化合物或肽跃迁。在一个实施方案中,与待测量蛋白质特异性结合的化合物是抗体或适体。在具体实施方案中,本文公开的诊断方法用于排除用于受试者的治疗方案,测量从受试者获得的样品中的蛋白质实验对象组的丰度,基于蛋白质测量计算癌症评分的概率,并且如果样品中确定的评分低于预定评分,则排除用于受试者的治疗方案。在一些实施方案中,实验对象组含有选自aldoa、fril、lg3bp、ibp3、lrp1、islr、tsp1、coia1、grp78、tetn、prdx1和cd14的至少3种蛋白质。任选地,实验对象组进一步包含选自ero1a、6pgd、gstp1、ggh、prdx1、cd14、ptpa、icam1、folh1、sodm、fiba、gslg1、rap2b或c163a的一种或多种蛋白质,或者选自lrp1、coia1、tsp1、aldoa、grp78、fril、lg3bp、bgh3、islr、prdx1、fiba或gslg的一种或多种蛋白质。在优选实施方案中,实验对象组含有至少tsp1、lg3bp、lrp1、aldoa和coia1。在更优选的实施方案中,实验对象组含有至少tsp1、lrp1、aldoa和coia1。在具体实施方案中,本文公开的诊断方法用于通过以下判定用于受试者的治疗方案:测量从受试者获得的样品中的蛋白质实验对象组的丰度,基于蛋白质测量计算癌症评分的概率,并且如果样品中确定的评分大于预定评分,则判定用于受试者的治疗方案。在一些实施方案中,实验对象组含有选自aldoa、fril、lg3bp、ibp3、lrp1、islr或tsp1或aldoa、fril、lg3bp、ibp3、lrp1、islr、tsp1、coia1、grp78、tetn、prdx1和cd14的至少3种蛋白质。任选地,实验对象组进一步包含选自ero1a、6pgd、gstp1、coia1、ggh、prdx1、sem3g、grp78、tetn、aifm1、mpri、tnf12、mmp9或ostp或coia1、tetn、grp78、apoe或tbb3的一种或多种蛋白质。在一些实施方案中,实验对象组包含lg3bp和c163a。在某些实施方案中,本文公开的诊断方法可以与其它临床评价方法组合使用,包括例如各种射线照相方法和/或侵入性方法。类似地,在某些实施方案中,本文公开的诊断方法可以用于鉴定用于其它临床评价方法的候选者,或评价受试者将获益于其它临床评价方法的可能性。生物样品如血浆或血清中某些蛋白质的高丰度可以阻碍分析目的蛋白质的能力,特别是在目的蛋白质以相对低的浓度表达的情况下。几种方法可用于避免这一问题,包括富集、分开和耗竭。富集使用亲和试剂按类别从样品中提取蛋白质,例如通过糖捕获去除糖基化蛋白质。分开使用方法如凝胶电泳或等电点聚焦,以将样品分成在蛋白质含量上在很大程度上不重叠的多重级分。耗竭通常通过利用使得能够去除大多数最丰富蛋白质的先进技术,例如igy14/supermix(sigmast.louis,mo),使用亲和柱来去除血液中最丰富的蛋白质,例如白蛋白。在本文提供的方法的某些实施方案中,在测定生物标记物或推定的生物标记物蛋白质表达水平之前,生物样品可以经受富集、分开和/或耗竭。在这些实施方案的某些中,血液蛋白质可以最初通过糖捕获法进行加工,所述糖捕获法富集糖基化蛋白质,允许定量测定以检测高pg/ml至低ng/ml浓度范围的蛋白质。糖捕获的示例性方法是本领域众所周知的(参见例如,美国专利号7,183,188;美国专利申请公开号2007/0099251;美国专利申请公开号2007/0202539;美国专利申请公开号2007/0269895;以及美国专利申请公开号2010/0279382)。在其它实施方案中,血液蛋白质可以最初通过蛋白质耗竭法进行加工,所述蛋白质耗竭法允许通过去除丰富蛋白质用于检测样品中通常模糊的生物标记物。在一个此类实施方案中,蛋白质耗竭法是supermix(sigma)耗竭法。在某些实施方案中,生物标记物蛋白质实验对象组包含2至100种生物标记物蛋白质。在这些实施例的某些中,实验对象组包含2至5、6至10、11至15、16至20、21-25、5至25、26至30、31至40、41至50、25至50、51至75、76至100种生物标记物蛋白质。在某些实施方案中,生物标记物蛋白质实验对象组包含生物标记物蛋白质的一个或多个子实验对象组,其各自包含至少两种生物标记物蛋白质。例如,生物标记物蛋白质实验对象组可以包含由在特定肺部状况下过表达的生物标记物蛋白质组成的第一子实验对象组、以及由在特定肺部状况下表达不足的生物标记物蛋白质组成的第二子实验对象组。在本文提供的方法、组合物和试剂盒的某些实施方案中,生物标记物蛋白质可以是与肺癌结合显示出差异表达的蛋白质。例如,在某些实施方案中,生物标记物蛋白质可以是表6中列出的与肺癌有关的蛋白质之一。在其它实施方案中,本文公开的诊断方法可以用于区分两种不同的肺部状况。例如,该方法可以用于将肺部状况分类为恶性肺癌相对于良性肺癌、nsclc相对于sclc、或肺癌相对于非癌性状况(例如炎性状况)。在某些实施方案中,提供了用于诊断受试者的肺部状况的试剂盒。这些试剂盒用于检测一种或多种生物标记物蛋白质的表达水平。任选地,试剂盒可以包含以标签或分开插页形式的使用说明书。试剂盒可以含有与本文所述的实验对象组中的蛋白质特异性结合的试剂。这些试剂可以包括抗体。试剂盒还可以含有与本文所述的实验对象组中的表达蛋白质的mrna特异性结合的试剂。这些试剂可以包括核苷酸探针。试剂盒还可以包括用于检测与本文所述的实验对象组中的蛋白质特异性结合的试剂的试剂。这些试剂可以包括荧光团。提供下述实施例以更好地说明本发明,并且不应解释为限制本发明的范围。就提到具体材料的程度而言,它仅仅出于说明的目的,并不预期限制本发明。本领域技术人员可以开发等价手段或反应物,而不需要发明能力的运用并且不脱离本发明的范围。实施例实施例1:肺癌生物标记物蛋白质的鉴定。回顾性病例对照研究设计用于鉴定生物标记物蛋白质及其实验对象组,用于诊断预定对照组和实验组中的各种肺部疾病。这些研究的第一个目标是证实关于对照组和实验组之间的个别蛋白质的统计学显著的差异表达。第二个目标是鉴定蛋白质实验对象组,其全部个别地证实对照组和实验组之间的统计学显著的差异表达。这个蛋白质实验对象组然后可以共同地用于区分二分疾病状态。具体的研究比较可以包括1)癌症相对于非癌症,2)小细胞肺癌相对于非小细胞肺癌(nsclc),3)癌症相对于炎性疾病状态(例如,传染性肉芽肿),或4)不同的结节尺寸,例如<10mm相对于≥10mm(取决于样品分布,可替代地使用10、15或20mm的截止)。关于每个受试者的数据由下述组成:来自先前入选机构审查委员会(institutereviewboard)(irb)批准研究的受试者的存档血浆样品用于鉴定生物标记物蛋白质和生物标记物实验对象组,其用于区分肺恶性肿瘤与非恶性肿瘤。血浆样品最初通过常规放血获得,等分且贮存于-80℃或更低温度下。样品制备,受试者标识代码分配,初始受试者记录输入和样本贮存按照irb研究方案执行。样品合格性基于临床参数,包括受试者、pn和临床分期参数。表7列出了用于纳入和排除的参数。表7纳入标准样品对于对照组或实验组的分配,及其进一步分层或与这些组内和这些组之间的其它样品的匹配,取决于关于受试者的各种临床数据。该数据包括例如人口统计学信息,例如年龄、性别和临床史(例如吸烟状况),共病状况,pn特征,以及切除病变和组织的病理学解释(表8)。表8研究设计和分析计划将表9中列出的对照:实验组配对区分优先次序。通过在实验组和对照组的分配中选择性包括表型例如吸烟效应,可以获得另外的临床和分子了解。经由病例对照分析中关于临床参数(例如年龄和结节大小)的样品分层或匹配,临床数据库中可用的人口统计学信息允许样品选择中的进一步改进。表9为实现蛋白质组学分析目的的实验组和对照组分配分析目的实验组对照组1区分癌症与良性肺部结节a.癌症结节具有直径≥4mm的结节的任何非恶性(良性)表型2区分癌症与非恶性(炎性、感染性)肺部结节a.癌症结节非恶性(非良性)肺部病症,例如肉芽肿(真菌)疾病,伴有结节执行lc-srm-ms以鉴定且定量血浆样品中的各种血浆蛋白质。在lc-srm-ms分析之前,每个样品使用igy14/supermix(sigma)进行耗竭,然后进行胰蛋白酶消化。将来自每个对照组或实验组的样品随机分批,并且在qtrap5500仪器(absciex,fostercity,ca)上一起加工,用于无偏比较。每次样品分析花费大约30分钟。对于所有肽和蛋白质收集并记录关于两个跃迁(天然和重标记)的峰面积。关于通过lc-srm-ms分析的每种蛋白质的数据输出通常产生由以下组成的四个测量:来自同一蛋白的两种肽各自的两个跃迁测量。这些测量允许推断靶蛋白的相对丰度,这用作其在生物信息学和统计分析中的表达水平。在对照组和实验组之间具有差异表达水平的生物标记物蛋白质的鉴定产生一种或多种新型的蛋白质组学谱。例如,鉴定了生物标记物蛋白质,其表达水平在被诊断为nsclc的具有pn的受试者相对于没有nsclc诊断的受试者中、或者在被诊断为nsclc相对于炎性病症的具有pn的受试者中不同。还鉴定了可以共同地区别二分疾病状态的生物标记物蛋白质实验对象组。可以适当地(先验地)操纵分析,以将1型和2型误差控制在0.05,并且检测25%的组群间差异/分析物。假设使用单侧配对非参数检验,一般评价个别蛋白质区分两个组群的诊断能力。这提供了关于证实实验组和对照组之间的差异表达所需的样本大小的下限。多重检验效应应用于鉴定用于评价诊断功效的蛋白质实验对象组,其需要更大的样本大小。用于确定关于个别蛋白质的差异表达的统计显著性的步骤顺序包括下述:1)评价并关联单个蛋白质跃迁的校准值(质量控制测量);2)使用曼怀二氏u检验(秩和)比较组的配对分析,以控制其它影响,以确定统计显著性;并且3)基于预定的显著性阈值,确定其显著性。跨越样品不关联(例如皮尔森相关性<0.5)的蛋白质内的跃迁视为不可靠的,并且从分析中排除。两个组群(例如癌症和非癌症)之间的校准样品比较,要求使用各种临床参数(例如结节大小、年龄和性别)的配对或匹配。此类配对控制了这些其它参数对实际比较目标(例如癌症和非癌症)的潜在影响。然后应用非参数检验例如曼怀二氏u检验(秩和),以测量组间的统计差异。可以使用多重检验校正(例如假发现率)来调整所得到的p值。置换检验可以用于进一步的显著性评价。显著性通过满足预定阈值(例如0.05)以滤出测定来确定,其中可能使用更高的阈值用于另外的过滤。另外的显著性标准是,三个重复测定中的两个必须个别地是显著的,以便使测定例如单个蛋白质是显著的。使用本文所述的统计方法鉴定蛋白质实验对象组,其个别地证实如上文定义的统计学显著的差异表达,并且可以共同地用于区分二分疾病状态。这要求开发多变量分类器,并且评价对于实验对象组的灵敏度、特异性和rocauc。另外,鉴定具有最佳区别性能,例如rocauc的蛋白质实验对象组,并且对于区别疾病状态的临床使用可能是足够的。用于确定蛋白质实验对象组的区别能力的统计显著性的步骤顺序包括:1)开发用于蛋白质实验对象组的多变量分类器,并且2)鉴定对于一组疾病状态具有最佳区别性能,例如rocauc的蛋白质实验对象组。开发用于蛋白质实验对象组的多变量分类器(例如,多数规则),包括视为重要的单一蛋白质测定。确定每个分类器的灵敏度和特异性,并且用于生成接受者操作特征(roc)曲线及其auc,以评价给定实验对象组对于特定比较(例如癌症相对于非癌症)的区别性能。方案1.审查来自呈现有肺部疾病的一组受试者的临床数据。2.提供来自受试者的血浆样品,其中所述样品是良性、癌性、copd或另一种肺部疾病。3.通过由结节的大小分开的pn,将良性或癌性的血浆样品分组。4.靶向371种推定的肺癌生物标记物蛋白质的库,所述蛋白质由至少两种肽/蛋白质和至少两种lc-srm-ms跃迁/肽组成。测量每个样本中的lc-srm-ms跃迁连同由10个跃迁组成的5个合成内部标准,以通过lc-srm-ms质谱法比较从血浆到合成内部标准的肽跃迁。5.定量每个跃迁的强度。6.将定量的跃迁针对内部标准进行标准化,以获得标准化强度。7.审查所测量的肽跃迁关于来自同一肽的关联性,拒绝不一致的跃迁。8.通过比较癌性与良性样品,对于每个跃迁生成roc。(roc比较特异性(真阳性)与(1-灵敏度)假阳性)。9.对于每个跃迁定义auc。(.5的auc是随机分类器;1.0是完美分类器)。10.确定auc截止点,以确定统计学显著的跃迁。11.定义超过auc截止点的跃迁。12.组合显著跃迁的所有配对。13.借助于逻辑回归,对于每个跃迁对定义新的auc。14.将配对组合重复成三元组、四元组等;定义基于合并跃迁的逻辑回归的新auc,直到已获得具有组合的所需性能(灵敏度和特异性)的生物标记物跃迁的实验对象组。15.针对先前未使用的血浆板实验对象组集合,验证了生物标记物跃迁的实验对象组。实施例2:使用生物标记物蛋白质的肺部疾病的诊断/分类。从呈现有pn的一个或多个受试者获得血浆样品,以评估受试者是否具有肺部状况。血浆样品使用igy14/supermix(sigma)进行耗竭,并且任选地经历一轮或多轮富集和/或分开,然后进行胰蛋白酶消化。使用lc-srm-ms测定,测量先前鉴定为在具有肺部状况的受试者中差异表达的一种或多种生物标记物蛋白质的表达水平。lc-srm-ms测定利用对于每种生物标记物蛋白质的2至5个肽跃迁。例如,该测定可以利用从表6中列出的任何蛋白质生成的一个或多个肽跃迁。如果一种或多种生物标记物蛋白质显示出的表达水平明显不同于该蛋白质的预定对照表达水平,则将受试者分类为具有肺部状况。实施例3:基于血液的诊断测试,以确定肺结节(pn)是良性或恶性的可能性。创建了15种蛋白质的实验对象组,其中这15种蛋白质的浓度相对于6种蛋白质标准的浓度指示癌症的可能性。使用质谱法方法学,测量了这15种蛋白质相对于6种蛋白质标准的相对浓度。分类算法用于将这些相对浓度合并成pn是良性还是恶性的相对可能性。进一步地,已证实存在关于这些实验对象组的许多变化,其也是pn是良性或恶性的可能性的诊断测试。本文描述了关于蛋白质实验对象组,蛋白质标准,测量方法学和/或分类算法的变化。研究设计开发了单反应监测(srm)质谱法(ms)测定,其由来自345种肺癌相关蛋白的1550个跃迁组成。上文描述了srm-ms测定和方法学。这项研究的目的是开发基于血液的诊断,用于将大小2cm以下的pn分为良性或恶性的。研究设计显示于表10中。表10.研究设计该研究由来自三个场所(laval、upenn和nyu)的242个血浆样品组成。表10中指示了来自每个场所的良性和恶性样品数目。该研究由来自其pn大小2cm或更小的患者的144个血浆样品、以及来自其pn大小大于2cm的患者的98个样品组成。这导致对于发现在大小为2cm或更小的良性和恶性癌症样品之间血液浓度为1.5倍或更高的蛋白质94%的估计效力。效力对于大小大于2cm的pn为74%。这项研究是回顾性多场所研究,其预期衍生对场所间变异稳固的肺癌的蛋白质生物标记物。该研究包括大于2cm的样品,以确保对于大小2cm或更小的肿瘤,由于测量技术(lc-srm-ms)的检测极限无法检测的蛋白质,在大小2cm或更大的肿瘤中仍可以检测到。来自每个场所和在每个大小类别(2cm以上和以下)中的样品关于结节大小、年龄和性别进行匹配。样品分析使用lc-srm-ms测量方法学,如下分析每个样品:1.使用来自sigma-aldrich的igy14和supermix耗竭柱,使样品耗竭高丰度蛋白质。2.使用胰蛋白酶将样品消化成胰蛋白酶肽。3.通过在waters纳米敏度lc系统上使用30分钟梯度的lc-srm-ms分析样品,随后为在ab-sciex5500三重四极杆装置上,1550个跃迁的srm-ms分析。4.获得原始的跃迁离子计数,并且对于1550个跃迁各自进行记录。必须指出,在每个步骤中,平行(步骤2和4)或连续背对背(步骤1和3)加工匹配样品。这使分析变异降到最低。最后,根据加工日,在样品的分批中执行样品分析的步骤1和2。如表10中所示,存在‘小’样品的五个分批和‘大’样品的四个分批。蛋白质候选清单跨越场所可再现地诊断的68种蛋白质的候选清单如下衍生。注意每种蛋白质都可以通过多重跃迁进行测量。步骤1:标准化鉴定了六种蛋白质,其具有在研究的所有样品中检测到的跃迁以及具有低变异系数。对于每种蛋白质,选择跨越样品具有最高中值强度的跃迁作为该蛋白质的代表性跃迁。这些蛋白质和跃迁在表11中可见。表11.标准化因子蛋白质(uniprotid)肽(氨基酸序列)跃迁(m/z)cd44_humanygfieghvvipr(seqidno:1)272.2tenx_humanyevtvvsvr(seqidno:2)759.5clus_humanassiidelfqdr(seqidno:3)565.3ibp3_humanflnvlspr(seqidno:4)685.4gels_humantasdfitk(seqidno:5)710.4masp1_humantgvitspdfpnpypk(seqidno:6)258.10我们将表11中的跃迁称为标准化因子(nf)。通过六个标准化因子中的每一个,将1550个跃迁中的每一个进行标准化,其中如下计算样品s中通过nff的跃迁t的新强度,表示为新(s,t,f):新(s,t,f)=原始(s,t)*中值(f)/原始(s,f)其中原始(s,t)是样品s中的跃迁t的原始强度;中值(f)是跨越所有样品的nff的中值强度;并且原始(s,f)是样品s中的nff的原始强度。对于每种蛋白质和标准化跃迁,计算每个分批的auc。选择使跨越9个分批的变异系数降到最低的nf作为该蛋白质和该蛋白质的所有跃迁的nf。因而,每一种蛋白质(及其所有跃迁)现在都由单个nf标准化。步骤2:可再现的诊断蛋白对于每个标准化跃迁,如下计算关于研究中的九个分批各自的其auc。如果在少于一半的癌症样品和少于一半的良性样品中检测到跃迁,则分批auc为‘nd’。否则,将分批中的良性和癌性样品进行比较,计算分批auc。对于每个跃迁,将分批auc值转换成百分位数auc评分。也就是说,如果标准化跃迁处于对于所有跃迁的auc评分的第82百分位数中,则对于该分批分配百分位数auc0.82。可再现的跃迁是满足下述标准中的至少一个的跃迁:1.在五个小分批的至少四个中,百分位数auc为75%或更高(或25%及更低)。2.在五个小分批的至少三个中,百分位数auc为80%或更高(或20%及更低),并且小分批中的剩余百分位数auc高于50%(低于50%)。3.在所有五个小分批中,百分位数auc高于50%(低于50%)。4.在四个大分批的至少三个中,百分位数auc为85%或更高(或15%及更低)。5.在四个大分批的至少三个中,百分位数auc为80%或更高(或20%及更低),并且大分批中的剩余百分位数auc高于50%(低于50%)。6.在所有四个大分批中,百分位数auc高于50%(低于50%)。这些标准导致具有满足一个或多个标准的至少一个跃迁的67种蛋白质的列表。这些蛋白质显示于表12中。步骤3:显著性和出现为了找到高性能实验对象组,执行了10,000次试验,其中在每次试验中,估计了选自表12的15种蛋白质的随机实验对象组的组合auc。为了计算15种蛋白质的每个实验对象组的组合auc,利用了最高强度的标准化跃迁。逻辑回归用于计算跨越所有小样品的15个实验对象组的auc。如图1中所示,15种蛋白质的131个实验对象组具有高于0.80的组合auc(通过研究分成小(<2.0cm)和大(>2.0cm)pn的显著性显示于图2中)。如图3中所示,尽管样品中基于场所的变化,实验对象组的弹性仍持续。表13中列出了实验对象组。为了计算15种蛋白质的每个实验对象组的组合auc,利用了最高强度的标准化跃迁。逻辑回归用于计算跨越所有小样品的15个实验对象组的auc。15种蛋白质的5个实验对象组具有高于0.80的组合auc。最后,表13中列出的131个实验对象组上的67种蛋白质各自的频率在表12中作为原始计数(第2列)和百分比(第3列)两者呈现。重要的观察是预先选择了15的实验对象组大小,以证明存在诊断蛋白和实验对象组。此外,存在众多此类实验对象组。也可以形成选自67种蛋白质的列表的较小实验对象组,并且可以使用此处相同的方法生成。实施例4:15种蛋白质的诊断实验对象组用于确定来自pn大小为2cm或更小的患者的血样是良性还是恶性的概率。在表14中,呈现了对所有小样品进行训练的逻辑回归分类器。表14.分类器具有以下结构其中c0和ci是对数回归系数,pi是对数转换的标准化跃迁强度。如果概率≥0.5,则样品被预测为癌症,否则为良性。在表14中,系数ci出现在第六列中,c0出现在最后一行中,并且关于每种蛋白质的标准化跃迁由第2列(蛋白质跃迁)和第4列(标准化因子)定义。作为roc图呈现的这种分类器的性能显示于图4中。总体auc为0.81。还可以通过将分类器个别地应用于每个研究场所来评价性能,其产生图5中出现的三个roc图。所得到的auc对于laval、nyu和upenn分别为0.79、0.88和0.78。实施例5:程序“ingenuity”®用于查询用于鉴定具有结节的患者中的肺癌的血液蛋白质,所述结节使用本发明的方法进行鉴定。使用来自被鉴定为诊断实验对象组(表13)的67种蛋白质的35种蛋白质(表15)的子集,执行了反向系统分析。查询了两个网络,其被鉴定为具有鉴定的35种蛋白质的癌症网络。结果显示,当在患者血液中发现的蛋白质查询直至核水平时,具有最高“命中”百分比的网络通过由尤其受香烟烟雾或肺癌调节的转录因子起始。还参见表16和图6。这些结果进一步证明了使用本发明的方法鉴定为用于肺癌诊断的蛋白质是预后且相关的。表15.表16.实施例6:用于诊断肺结节的协同蛋白。为了实现协同蛋白的无偏发现,利用了选择性反应监测(srm)质谱法(addona,abbatiello等人2009)。srm是质谱法的一种形式,其监测选定蛋白质的特定信息性(蛋白质型(proteotypic))肽的预定和高特异性质量产物。这些肽被识别为质谱中的特异性跃迁。srm具有其它技术(尤其是基于抗体的技术)不具备的下述所需特点:●可以快速且成本效益地开发用于数十种或数百种蛋白质的高度复用srm测定。●开发的测定用于个人选择的蛋白质,并不局限于现有测定的目录。此外,可以对于蛋白质的特异性区域,例如肿瘤细胞的细胞表面上的跨膜蛋白质的细胞外部分,或对于特异性同种型开发测定。●从发现到临床测试均可使用srm技术。肽离子化,质谱法的基础,是显著地可再现的。使用单一技术平台避免了将测定从一种技术平台转移到另一种的常见问题。srm多年来一直用于小分子分析物的临床测试,并且最近还用于生物学相关测定的开发[10]。标记的和未标记的srm肽是商购可得的,连同用于设计和进行srm分析的质谱的开放源文库和数据储库。存在特别的公共资源,以加速测定开发,包括peptideatlas[11]和plasmaproteomeproject[12、13],srmatlas和passel,peptideatlassrmexperimentallibrary(www.systemsbiology.org/passel)。引入了增强技术性能的两种srm策略。首先,大规模srm测定开发引入了监测假信号的可能性。使用表达相关技术的扩展[14],假信号监测率降低到3%以下。这与由mprophet所使用的方法(reiter,rinner等人,2011)是可比较和互补的。其次,内源蛋白质的实验对象组用于标准化。然而,尽管通常选择这些蛋白质作为“管家”蛋白质(lange,picotti等人,2008),但鉴定了对于该技术平台为强标准化物的蛋白质。也就是说,蛋白质监测技术变化的效应,使得它可以有效地加以控制。例如,这导致由于高丰度蛋白质的样品耗竭,从23.8%到9.0%的技术差化降低。先前已讨论了内源信号标准化的益处(price,trent等人,2007)。该策略的最后组分是使用新兴的最佳实践,小心地设计发现研究和验证研究。具体地,病例(恶性结节)和对照(良性结节)在年龄、结节大小、性别和参与的临床场所方面是成对匹配的。这确保发现的候选标记物不是年龄的标记物或在样品收集中的场所间变化。研究是功能强大的,包括多重场所,参与验证研究的新场所,并且重要的是,设计为解决测试的预期用途。样品的小心选择和匹配导致了分类器格外有价值的特点。分类器生成不依赖于结节大小和吸烟状态的评分。由于这些是目前用于ipn的临床管理的危险因素,因此分类器是用于ipn的诊断中的补充性分子工具。用于测定开发的生物标记物候选物选择为了鉴定血液中源于肺肿瘤细胞的肺癌生物标记物,获得了切除的肺肿瘤和相同叶的远侧正常组织。质膜从内皮细胞和上皮细胞两者中分离,并且通过串联质谱法分析,以鉴定在肿瘤细胞上过表达的细胞表面蛋白。类似地,分离高尔基体,以鉴定来自肿瘤细胞的过度分泌的蛋白质。有证据存在于血液中或分泌的蛋白质被区分优先次序,导致217种蛋白质的集合。关于细节,参见实施例7:材料和方法。为了确保其它可行的肺癌生物标记物不被忽略,执行了文献检索并且手动精选肺癌标记物。如上文,有证据存在于血液中或分泌的蛋白质被区分优先次序。这导致319种蛋白质的集合。关于细节,参见实施例7:材料和方法。组织(217)和文献(319)候选物重叠了148种蛋白质,导致388种蛋白质候选物的最终候选物列表。参见实施例7:材料和方法。srm测定的开发使用标准合成肽技术开发了用于388种蛋白质的srm测定(参见实施例7:材料和方法)。在388种候选物中,对于371种候选物成功开发了srm测定。将371个srm测定应用于良性和肺癌血浆样品,以评估血液中的检测率。检测到190个(51%成功率)srm测定。这一成功率与类似尝试有利地比较,以开发大规模srm测定用于检测血浆中的癌症标记物。最近,从1172种候选物中开发了用于一般癌症标记物的182个srm测定(16%成功率)[15]。尽管仅集中于肺癌标记物,但效率中的3倍增加很可能是由于从具有存在于血液中的先前证据的癌症组织中寻找候选物来源。在血液中通过质谱法先前检测到的371种蛋白质具有在血液中64%的检测成功率,而没有的蛋白质具有35%的成功率。在血液中检测到的190种蛋白质中,114种衍生自组织来源的候选物,而167种衍生自文献来源的候选物(91种蛋白质重叠)。参见实施例7:材料和方法以及表6。通常,手动精选srm测定,以确保测定监测预期的肽。然而,这对于大规模srm测定(例如这种371蛋白测定)变得不可行。最近以来,计算工具如mprophet(reiter,rinner等人,2011)使得srm测定的自动化合格成为可能。引入了对mprophet的补充策略,其不要求对于每个数据集的定制。它利用了相关技术(kearney,butler等人,2008),以高置信度确认蛋白质跃迁的身份。在图7中,呈现了测定中的每一对跃迁之间的皮尔森相关性的直方图。在下文详述的发现研究中,一对跃迁之间的相关性得自其在所有143个样品中的表达谱。如期望的,来自相同肽的跃迁是高度相关联的。类似地,来自相同蛋白质的不同肽片段的跃迁也是高度相关联的。相比之下,来自不同蛋白质的跃迁并非高度相关联的,并且使得蛋白质的srm测定质量的统计分析成为可能。例如,如果来自相同蛋白质的两种肽的跃迁的相关性高于0.5,则存在该测定为假的小于3%的概率。参见实施例7:材料和方法。分类器发现表17中显示了用于分类器发现的143个样品的概括。从三个场所获得样品,以避免对单个场所的过度拟合。参与场所为laval(institutuniversitairedecardiologieetdepneumologiedequebec)、nyu(newyorkuniversity)和upenn(universityofpennsylvania)。还根据结节大小(直径)、年龄和吸烟状况,选择了代表预期用途群体的样品。通过在年龄、性别、场所和结节大小方面的匹配,来配对良性样品和癌症样品(良性样品和癌症样品要求具有放射学鉴定的结节)。良性样品和癌症样品展示在吸烟(包年)方面的偏差,然而,大多数良性样品和癌症样品是当前或过去的吸烟者。在比较恶性和良性样品时,意图是发现其为肺癌标记物;而不是年龄、结节大小或场所样品收集中的差异的标记物的蛋白质。注意癌症样品得到病理学确认,而良性样品得到病理学或放射学确认(在两年的ct扫描监测中未证实肿瘤生长)。表17:用于发现集和验证集的临床数据概括和人口统计学分析。样品的加工是分批进行的。每个分批含有一组随机选择的癌症-良性对,以及包括用于校准和质量控制的目的的三个血浆标准。所有血浆样品都进行免疫耗竭,胰蛋白酶消化,并且通过反相hplc-srm-ms进行分析。使用内源蛋白质实验对象组,对蛋白质跃迁进行标准化。标准化程序设计为降低总体变化性,但特别是降低通过耗竭步骤引入的变化性。总体技术变化性从32.3%降低到25.1%,并且由于耗竭的技术变化性从23.8%降低到9.0%。样品分析和标准化程序的细节在实施例7:材料和方法中可获得。为了评价蛋白质实验对象组,使它们与逻辑回归模型拟合。选择逻辑回归以避免用非线性模型可能发生的过拟合,尤其是当所测量的变量(跃迁)数目与研究中的样品数目相似或更大时。实验对象组的性能通过部分曲线下面积(auc)进行测量,其中灵敏度固定在90%(mcclish1989)。部分auc与高npv性能相关联,同时使ror达到最大。为了衍生13蛋白质分类器,使用了四个标准:●蛋白质必须具有跨越研究中的样品可靠地检测到高于噪声的跃迁。●蛋白质必须是高度协同的。●蛋白质必须具有稳固的跃迁(高信噪比,无干扰等)●在逻辑回归模型内,蛋白质的系数在交叉验证期间必须具有较低变化性,即,它必须是稳定的。这些标准各自如何应用的细节显示于实施例7:材料和方法中。最后,通过蒙特卡洛交叉验证(mccv),将13蛋白质分类器对逻辑回归模型进行训练,具有20%的留出率和20,000次迭代。表18中列出了用于排除分类器的13种蛋白质,连同其最高强度跃迁和模型系数。表18:13蛋白质分类器。排除分类器的验证52个癌症样品和52个良性样品(参见表17)用于验证13蛋白质分类器的性能。所有样品都不依赖于发现样品,另外,超过36%的验证样品来源自新的第四个场所(vanderbiltuniversity)。样品选择为与预期用途一致,并且根据性别、临床场所和结节大小进行匹配。我们注意到轻微的年龄偏差,这是由于来自年轻患者的5个良性样品。预计90%的npv,95%置信区间为+/-5%。至此,我们指在143个样品上训练的13蛋白质分类器,发现分类器。然而,一旦完成验证以找到用于分类器的最佳系数,它就对所有247个样品(发现集和验证集)进行重新训练,因为这最预测未来的性能。我们将这种分类器称为最终分类器。最终分类器的系数显示于表21中。在图8中概括了发现分类器和最终分类器的性能。报告的是当应用于发现集、验证集时,关于发现分类器的npv和ror。对于所有样品、以及结节大小局限于8mm至20mm的所有样品(191个样品),报告了关于最终分类器的npv和ror。npv和ror各自报告为0至1的分数。类似地,分类器产生0至1之间的评分,这是由分类器预测的癌症概率。关于npv和ror的发现和验证曲线与所期望的优越的发现曲线相似。这证实了对独立样品集的性能可再现性。对于发现样品和验证样品,排除阈值为0.40的发现分类器分别达到96%和90%的npv,而ror分别为33%和23%。对于所有样品以及局限于8mm-20mm的所有样品,排除阈值为0.60的最终分类器分别达到91%和90%的npv,而ror分别为45%和43%。分类器的应用图9呈现了最终分类器对来自发现集和验证集的所有247个样品的应用。图9的意图是将吸烟(以包年测量)和结节大小(与每个圆圈的大小成比例)的临床危险因素与分配给每个样品的分类器评分形成对照。首先,注意具有高分类器评分的癌症样品的密度。该分类器已设计为以高灵敏度检测血液中的癌症标记。结果,假设25%的癌症流行率,在排除阈值(0.60)的左侧,存在极少数(<10%)的癌症样品[16,17]。第三是结节大小似乎没有随着分类器评分而增加的观察。大结节和小结节两者均在分类器评分谱中分散。类似地,尽管存在具有高分类器评分的一些非常重的吸烟者,但吸烟的增加看起来没有随着分类器评分而增加。为了定量这一观察,计算了分类器评分与结节大小、吸烟和年龄之间的相关性,并且显示于表19中。在所有情况下,分类器评分与危险因素之间不存在显著关系。一个例外是良性分类器评分与良性年龄之间的弱相关性。然而,这种相关性如此弱,使得分类器评分每10年仅增加0.04。年龄结节大小吸烟良性0.25-0.060.11癌症0.01-0.010.06表19:分类器评分与临床危险因素之间的相关性。这种相关性的缺乏具有临床实用性。它暗示分类器提供了关于ipn的疾病状态的分子信息,其随风险因素(例如结节大小和吸烟状态)而增加。因而,它是医生针对ipn的临床管理做出更明智决定的临床工具。为了显现这可能是如何实现的,我们演示了通过分类器生成的癌症概率评分如何可以与癌症风险相关(参见图11)在给定的分类器评分下,所有癌症结节的一定百分比将具有较小的评分。这是分类器的灵敏度。例如,在分类器评分0.8下,47%的癌症患者具有较低的评分,在分类器评分0.7下,28%的癌症患者具有较低的评分;在分类器评分0.5下,仅9%是较低的,且最后在评分0.25下,仅4%是较低的。这使得医生能够根据相对风险来解释患者的分类器评分。分类器的分子基础目标是通过选择通过srm协同的、稳固检测并且在分类器内稳定的蛋白质,来鉴定恶性肺结节的分子标记。衍生分类器与肺癌如何良好地关联是否存在关于血液中的这13种蛋白质的扰动的分子基础并且最后,分类器在其它可能的蛋白质组合中有何独特之处为了回答这些问题,将分类器的13种蛋白质提交,用于使用ipa(ingenuitysystems,www.ingenuity.com)的途径分析。第一步是从细胞外部向内部进行工作,以鉴定最可能引起这13种蛋白质的调节的转录因子。最重要的五种是fos、nrf2、ahr、hd和myc。fos是许多形式的癌症所共有的。然而,nrf2和ahr与肺癌、对氧化性应激的应答和肺部炎症有关。myc与肺癌以及对氧化性应激的应答有关,而hd与肺部炎症以及对氧化性应激的应答有关。13种分类器蛋白对这三个网络(肺癌、对氧化性应激的应答和肺部炎症)也是高度特异性的。这概括于图10中,其中描绘了分类器蛋白(绿色)、转录因子(蓝色)和三个合并的网络(橙色)。尽管只有islr没有通过这三个肺特异性网络与其它蛋白质连接,但它通过对癌症非特异性的癌症网络连接。总之,13种分类器蛋白的调节可以追溯到对肺癌、肺部炎症和氧化性应激网络特定性的几种转录因子。为了解决分类器唯一性的问题,形成了来自21种稳固和协同的蛋白质的每一个分类器(表20)。由于计算量过大,这些分类器无法通过蒙特卡洛交叉验证进行充分训练,因而,只能获得其性能的估计值。鉴定了五个高性能替代分类器,然后进行充分训练。表20中显示了分类器和五个高性能替代物。每种蛋白质的频率显示于计数列中,特别地,前11种蛋白质出现在6个分类器的4个中。这11种蛋白质具有比剩余蛋白质明显更高的协同评分。通过这种分析,似乎存在形成恶性结节的血液标记的蛋白质核心组。表20:分类器和高性能替代物;显示了在分别的实验对象组上关于蛋白质的系数。这一结果提示存在限定高性能分类器的蛋白质核心组,但存在替代实验对象组。然而,实验对象组成员资格中的改变影响npv和ror之间的权衡。实施例7:材料和方法。来源自组织的测定开发候选物在irb的批准下以及伴随患者知情同意书,从centrehospitalierdel’universitédemontréalandmcgilluniversityhealthcentre收集了得自新鲜肺肿瘤切除术的患者样品。样品得自肿瘤以及同一肺叶的远侧正常组织。然后使用免疫亲和力方案,从30个患者(19个腺癌、6个鳞状癌、5个大细胞癌)的上皮细胞和38个患者(13个腺癌、18个鳞状癌、7个大细胞癌)的内皮细胞中分离每对样品的质膜。使用等密度离心,随后为碳酸铵提取,从来自33个患者(18个腺癌、14个鳞状癌、1个腺鳞癌)的每对样品中分离高尔基体。然后通过串联质谱法分析质膜分离物和高尔基体分离物,以鉴定对于质膜和高尔基体两者,在肺癌组织中过表达超过正常组织的蛋白质。来源自文献的测定开发候选物从两个公共数据库和一个商业数据库中鉴定候选肺癌生物标记物:entrez、nbk3836、uniprot和nextbio。对于数据库查询预定义了术语,所述数据库查询使用perl脚本自动化。分别于2010年5月6日(uniprot)、2010年5月17日(entrez)和2010年7月8日(nextbio)进行数据挖掘。然后将生物标记物组装并映射到uniprot鉴定器。在血液中存在的证据组织来源和文献来源的生物标记物候选物要求具有在血液中存在的证据。对于通过质谱法检测的证据,使用了三个数据集。hupo9504含有通过串联质谱法鉴定的9504种人蛋白[13]。hupo889hupo889,hupo9504的较高置信度子集,含有889种人蛋白[18]。还使用了peptideatlas(2009年11月构建)。如果生物标记物候选物含有至少一种hupo889、或至少两种hupo9504肽、或至少两种peptideatlas肽,则它被标记为先前检测到的。除通过质谱法在血液中检测的直接证据之外,作为分泌性蛋白或单次跨膜蛋白[19]的注释也接受为在血液中存在的证据。此外,uniprot中的蛋白质或指名为血浆蛋白质,使用用于预测蛋白质是否分泌到血液内的三种程序。这些程序是tmhmm[20]、signalp[21]和secretomep[22]。如果tmhmm预测蛋白质具有一个跨膜结构域,而signalp预测该跨膜结构域被切割;或tmhmm预测蛋白质没有跨膜结构域,而signalp或secretomep预测蛋白质被分泌,则该蛋白质被预测为分泌的。srm测定开发使用类似于文献[15、23、24]中描述的方案,基于合成肽开发了用于388种靶向蛋白的srm测定。从公共来源例如peptideatlas,humanplasmaproteomedatabase,或通过蛋白质型预测工具[25]鉴定了至多5种srm合适的肽/蛋白质,并且进行合成。对于双重荷电和三重荷电的前体离子,在absciex5500qtrap上收集srm触发的ms/ms波谱。使用mascot(截止评分≥15),将获得的ms/ms波谱分配给个别肽[26]。选择至多四个跃迁/前体离子用于优化。所得到的相应的最佳保留时间、去簇电压和碰撞能对于所有跃迁进行组装。对所有合成肽的混合物、良性患者的合并样品和癌症患者的合并样品测量了最佳跃迁。分批分析跃迁,每个分批含有至多1750个跃迁。两种生物样品均进行免疫耗竭,并通过胰蛋白酶进行消化,并且在与反相(rp)高效液相层析(hplc)系统偶联的absciex5500qtrap上进行分析。手动审查获得的srm数据,以选择两种最佳的肽/蛋白质和两个最佳的跃迁/肽。不选择具有由其它跃迁的干扰的跃迁。合成肽混合物中肽的两个最佳跃迁的强度之间的比率,也用于评价生物样品中跃迁的特异性。强度比被视为定义srm测定的重要度量。血浆样品的加工使用填充有来自sigma的igy14-supermix树脂的免疫耗竭柱,序贯地耗竭血浆样品的高丰度蛋白质和中等丰度蛋白质。然后将耗竭的血浆样品变性,通过胰蛋白酶消化并脱盐。使用毛细管反相lc柱(thermobiobasic18kappa;柱尺寸:320μmx150mm;粒度:5μm;孔径:300å)和纳米hplc系统(nanoacquity,watersinc.),将肽样品分开。流动相为(a)0.2%甲酸的水溶液和(b)0.2%甲酸的乙腈溶液。将样品注入(8μl),并且使用线性梯度(经过19分钟,98%a至70%a,5μl/分钟)分开。肽被直接洗脱到质谱仪(5500qtraplc/ms/ms,absciex)的电喷雾源内,所述质谱仪以预定的srm正离子模式运行(q1分辨率:单位;q3分辨率:单位;检测窗口:180秒;循环时间:1.5秒)。然后通过软件multiquant(absciex)整合跃迁强度。10,000的强度阈值用于过滤出噪声数据和未检测到的跃迁。用于发现研究和验证研究的血浆样品血浆样品的等分试样由institutuniversitairedecardiologieetdepneumologiedequebec(iucpq,hospitallaval)、newyorkuniversity、universityofpennsylvania和vanderbiltuniversity提供(参见表17)。受试者分别入选到先前由其伦理审查委员会(ethicsreviewboard)(erb)或机构审查委员会(irb)批准的临床研究中。另外,根据需要由相关机构的irb审查并批准申办者的研究方案后,由研究人员提供血浆样品。用于蛋白质组学分析的样品合格性基于研究纳入标准和排除标准的满足,包括受试者的人口统计学信息、受试者通过胸部计算机断层摄影(ct)的相应肺部结节射线照相特征、以及在诊断性手术切除时获得的肺部结节的组织病理学。癌症样品具有非小细胞肺癌(nsclc)(包括腺癌、鳞状细胞癌、大细胞癌或支气管肺泡细胞癌)的组织病理学诊断,以及30mm或更小的射线照相结节。良性样品包括肉芽肿、错构瘤和疤痕组织,也要求具有30mm或更小的射线照相结节,以及与临床指南一致的非恶性的组织病理学确认或放射学确认。为了确保临床数据的准确度,根据由1996年的健康保险携带和责任法案(healthinsuranceportabilityandaccountabilityact)(hipaa)制定的指导,执行与受试者和肺部结节两者相关的临床数据的独立监测和验证,以确保受试者隐私权。研究设计研究设计的目的是消除临床和技术偏差。在临床上,将癌症样品和良性样品配对,使得它们来自相同的场所、相同的性别、在10mm内的结节大小,在10年内的年龄、以及在20包年内的吸烟史。至多15对匹配的癌症样品和良性样品/分批迭代分配给加工分批,直到基于年龄、性别或结节大小无法证实统计偏差。进一步将每个加工分批内的配对样品随机且重复地分配至加工分批内的位置,直到位置与性别、结节大小和年龄之间的相应皮尔森相关系数的绝对值小于0.1。其后,将每对癌症样品和良性样品随机分配到其相对位置。为了提供用于样品分批的对照,将合并的人血浆标准(hps)(bioreclamation,hicksville,ny)的三个200µl等分试样分别放置在每个加工分批的开始、中间和结尾。分批内的样品一起进行分析。逻辑回归模型逻辑回归分类方法[27]用于将跃迁实验对象组组合到分类器内,并且对于每个样品计算在0和1之间的分类概率评分。样品的概率评分确定为其中是样品s中对数转换(底数2)的标准化的跃迁强度,是相应的对数回归系数,是分类器特异性常数,并且是分类器中的跃迁总数目。如果小于决策阈值,则样品被分类为良性的。决策阈值可以根据所需的npv来增加或减少。为了定义分类器,必须从发现研究学习(即训练)跃迁(即蛋白质)实验对象组、其系数、标准化跃迁、分类器系数和决策阈值,然后使用验证研究进行确认。排除分类器的发现用于分类器发现的143个样品的概括显示于表17中,并且如上所述进行加工。如上所述将蛋白质跃迁标准化。消除了在至少50%的癌症样品或50%的良性样品中未检测到的跃迁,留下117个跃迁用于进一步考虑。关于这些跃迁的缺失值被替换为对于该跃迁在所有样品中的最小检测值的一半。下一步是找到最协同蛋白的集合。蛋白质的协同评分是其参与的高性能实验对象组数目除以它可以单独偶然出现的此类实验对象组数目。因此,高于1的协同评分是良好的,而低于1的评分则不是。通过下述程序估计关于每种蛋白质的协同评分:生成了选自117种候选物、各10种蛋白质的一百万个随机实验对象组。使用蒙特卡洛交叉验证(mccv)方法,以20%的留出率和一百种样品排列/实验对象组),对10种蛋白质的每个实验对象组进行训练,以拟合逻辑回归模型,并且通过部分auc评价其性能[28]。通过生成如此大量的实验对象组,我们足够良好地对分类器的空间进行取样,以偶然地找到一些高性能者。保留了所生成的一百万个中的一百个最佳随机实验对象组(参见表2),并且对于117种蛋白质中的每一种,我们确定了每种蛋白质如何频繁地出现在这些顶部实验对象组上。在去除内源性标准化物后,在117种蛋白质中,36种具有比偶然预期的更高的频率。(表22)蛋白质偶然出现在其上的预期实验对象组数目为100*10/117=8.33。蛋白质的协同评分是其出现的实验对象组数目除以8.33。通过手动审查原始srm数据,并且消除由于低信噪比或干扰而没有稳固srm跃迁的蛋白质,将36种协同蛋白的集合进一步减少为21种蛋白质的集合。从21种蛋白质的集合中迭代消除蛋白质,直到获得具有最佳部分auc的分类器。用于消除的标准是系数稳定性。在逻辑回归模型中,每种蛋白质都具有系数。在训练模型的过程中,确定每种蛋白质的系数。当这使用交叉验证(mccv)执行时,衍生关于每种蛋白质的数百个系数估计值。这些系数的变化性是蛋白质稳定性的估计。在每个步骤中,使用mccv(留出率20%,一万个样品排列/实验对象组),将蛋白质对逻辑回归模型进行训练,并且测量其稳定性。消除了最不稳定的蛋白质。这一过程继续直至达到具有最佳部分auc的13蛋白质分类器。最后,通过mccv(留出率20%,两万个样品排列),将13蛋白质分类器对逻辑回归模型进行训练。表18列出了用于排除分类器的13种蛋白质,连同其最高强度跃迁和模型系数。决策阈值的选择假设肺结节的癌症流行率为,如下根据灵敏度()和特异性()计算分类器(npv和ror)对具有肺部结节的患者群体的性能:。然后选择对于癌症或良性样品分开调用的阈值作为npv90%和ror20%的概率评分。由于我们预期在发现集上测量的分类器性能是高估的,因此将阈值选择为范围,因为性能在独立的验证集上通常下降。排除分类器的验证52个癌症样品和52个良性样品(参见表17)用于验证13蛋白质分类器的性能。将一半样品放入在发现样品后立即进行分析的预定加工分批中,而另一半样品在以后的日期进行分析。这引入在实践中将预期的变化性。更具体而言,将每个加工分批中运行的三个hps样品用作外部校准物。关于hps校准的细节在下文描述。通过hps样品的校准对于无标记的ms方法,预期关于在不同实验之间的信号强度的变化。为了减少这种变化,我们利用hps样品作为外部标准,并且在发现研究和验证研究之间校准了强度。假设是样品s中对数转换(底数2)的标准化的跃迁强度,和分别是发现研究和验证研究中hps样品的相应中值。然后,hps校正强度为因而,假设验证研究中关于临床样品的癌症概率由分类器预测为。然后,如下计算hps校正的临床样品的癌症概率:其中和。此处,和分别是发现研究和验证研究中所有hps样品的的中值。统计分析所有统计分析都用stata、r和/或matlab执行。耗竭柱漂移随着越来越多的样品被同一柱耗竭,我们观察到信号强度的增加。我们使用hps样品中的跃迁强度来定量这种技术变化性。假设是hps样品的跃迁强度,样品的漂移定义为其中是由同一柱耗竭的所有hps样品中的平均值,并且中值取自样品中所有检测到的跃迁。然后,柱的漂移定义为。此处,中值取自由柱耗竭的所有hps样品。如果样品漂移不大于或小于零,则相应的中值视为0。中值柱漂移是研究中使用的所有耗竭柱的漂移中值。内源标准化蛋白质的鉴定下述标准用于将跃迁鉴定为标准化物:●具有来自同一蛋白质的所有跃迁的最高中值强度。●在所有样品中都检测到。●作为标准化物在降低中值技术cv(在hps样品上测量的跃迁强度的中值cv)方面排名很高。●在降低样品耗竭中观察到的中值层析柱漂移方面排名很高。●具有较低的中值技术cv和较低的中值生物学cv(在临床样品上测量的跃迁强度的中值cv)。选择了六个跃迁,并且显示于表23中。标准化物跃迁seqidno中值技术cv(%)中值柱漂移(%)pedf_humanlqslfdspdfsk_692.34_593.302825.86.8masp1_humantgvitspdfpnpypk_816.92_258.10626.518.3gels_humantasdfitk_441.73_710.40527.116.8lum_humansledlqlthnk_433.23_499.302927.116.1c163a_humaninpasldk_429.24_630.303026.614.6ptprj_humanvitepipvsdlr_669.89_896.503127.218.2通过跃迁的实验对象组标准化25.19.0无需标准化32.323.8表23:内源标准化物的实验对象组。数据标准化六个标准化跃迁的实验对象组(参见表23)用于标准化原始srm数据,用于两个目的:(a)降低同一研究内的样品间强度变化,以及(b)降低不同研究之间的强度变化。对于第一个目的,对于每种样品计算比例因子,使得样品的六个标准化跃迁的强度与所有hgs样品的相应中值强度保持一致。假设是样品s中标准化跃迁的强度,并且是所有hgs样品的相应中值强度,则关于样品的比例因子由给出,其中是强度比的中值,并且是研究中所有样品的中值。对于第二个目的,比例因子在发现研究和验证研究之间进行计算,使得验证研究中所有hgs样品的六个标准化跃迁的中值强度与发现研究中的相应值可比较。假设两项研究中所有hgs样品的中值强度分别为和,则关于验证研究的比例因子由以下给出最后,对于每种样品的每个跃迁,其标准化强度计算为:其中是原始强度。从组织中分离膜蛋白从得自新鲜肺切除物的正常和肿瘤肺组织样品中分离内皮细胞质膜蛋白。简言之,在缓冲液中洗涤组织,并且通过用polytron破坏组织来制备匀浆物。匀浆物通过180-μm筛孔进行过滤,并且滤液在4℃下以900xg离心10分钟。将上清液在4℃下以218,000xg在50%(w:v)蔗糖垫的顶部上离心60分钟,以使膜形成团块。将团块重悬浮并用微球菌核酸酶处理。使来自内皮细胞的膜与抗血栓调节蛋白、抗ace、抗cd34和抗cd144抗体的组合一起温育,然后在4℃下以280,000xg在50%(w:v)蔗糖垫的顶部上离心60分钟。在将团块重悬浮后,使用macs微珠分离内皮细胞质膜,并且用碘化钾处理以去除细胞质外周蛋白。从新鲜肺切除物中分离来自正常和肿瘤肺组织样品的上皮细胞质膜蛋白。如上文对于内皮细胞质膜蛋白制备所述洗涤组织并匀浆化。来自上皮细胞的膜用抗esa、抗cea、抗cd66c和抗ema抗体的组合进行标记,然后在4℃下以218,000xg在50%(w:v)蔗糖垫的顶部上离心60分钟。使用macs微珠分离上皮细胞质膜,并且将洗脱物在4℃下以33,700xg在33%(w:v)蔗糖垫上离心30分钟。在去除上清液和蔗糖垫后,将团块重悬浮于laemmli/尿素/dtt中。从组织中分离分泌性蛋白从分离自新鲜肺切除物的正常和肿瘤肺组织样品中分离分泌性蛋白。洗涤组织,并且使用polytron匀浆进行匀浆化。用浓缩蔗糖将匀浆物的密度调整至1.4m,然后通过在0.8和1.2m的不连续蔗糖梯度上,在4℃下以100,000xg等密度离心2小时来分离分泌囊泡。收集在0.8/1.2m界面处浓缩的囊泡,并且与0.5mkcl(最终浓度)进一步温育25分钟,以去除松散结合的外周蛋白。通过在4℃下以150,000xg超速离心一小时来回收囊泡,然后在4℃下用100mm碳酸铵ph11.0打开30分钟。在4℃下以150,000xg超速离心1小时之后,从上清液中回收分泌性蛋白。igy14-supermix免疫亲和柱的制备使用分别含有2:1比率的igy14和supermix免疫亲和树脂(sigmaaldrich)的浆料,内部制备免疫亲和柱。简言之,将混合的免疫亲和树脂的浆料(10ml,50%)加入玻璃层析柱(tricorn,gehealthcare)中,并且允许树脂在重力流动下沉降,导致柱中的5ml树脂体积。将柱加盖并且放置在agilent1100系列hplc系统上,用于进一步填充(20分钟,0.15m碳酸氢铵,2ml/分钟)。然后,通过重复注射hps样品的等分试样,评价研究中使用的每个柱的性能。在开始临床样品的每个分批的免疫亲和分离之前评价柱性能。igy14-sumermix免疫亲和层析在免疫亲和分离之前,将血浆样品(60μl)稀释(分别为0.15m碳酸氢铵,1:2v/v)并过滤(0.2μmacroprep96孔滤板,palllifesciences)。使用三种缓冲液(上样/洗涤:0.15m碳酸氢铵;汽提/洗脱:0.1m甘氨酸,ph2.5;中和:0.01mtris-hcl,0.15mnacl,ph7.4),在连接到agilent1100系列hplc系统的igy14-supermix柱上,用上样-洗涤-洗脱-中和-再平衡循环(总时间为36分钟),来分离稀释血浆(90μl)。未结合级分和结合级分使用uv吸光度(280nm)进行监测,并且在分离后分辨基线。仅收集含有低丰度蛋白质的未结合级分,用于下游加工和分析。未结合级分在酶促消化之前进行冻干。低丰度蛋白质的酶促消化低丰度蛋白在温和的变性条件下(200μl的1:10.1m碳酸氢铵/三氟乙醇v/v)重构,并且允许温育(30分钟,室温,定轨振荡器)。然后将样品稀释(800μl0.1m碳酸氢铵),并且用胰蛋白酶消化(princetonseparations;0.4μg胰蛋白酶/样品,37℃,16小时)。消化的样品在固相提取之前进行冻干。固相提取在质谱法之前,使用固相提取来降低样品中的盐和缓冲液含量。将含有胰蛋白酶肽的冻干样品重构(350μl0.01m碳酸氢铵),并且允许温育(15分钟,室温,定轨振荡器)。然后将还原剂加入样品中(30μl0.05mtcep),并且使样品温育(60分钟,室温)。加入稀酸和低百分比的有机溶剂(375μl90%水/10%乙腈/0.2%三氟乙酸),以优化肽的固相提取。提取板(emporec18,3mbioanalyticaltechnologies)根据制造商方案进行条件化。将样品加载到固相提取板上,洗涤(500μl95%水/5%乙腈/0.1%三氟乙酸),并洗脱(200μl52%水/48%乙腈/0.1%三氟乙酸)到收集板内。将洗脱物拆分成两个相等的等分试样,并且将每个等分试样在真空浓缩器中干燥。一个等分试样立即用于质谱法,而另一等分试样被贮存(-80℃)并根据需要使用。紧在lc-srmms分析之前,重构样品(12μl90%水/10%乙腈/0.2%甲酸)。纳入标准和排除标准如果血浆样品(a)在edta管中获得,(b)得自在参与机构先前入选irb批准的研究的受试者,以及(c)存档的,例如如通过研究方案要求的标记、等分并冷冻的,则它们对于研究是合格的。样品还必须满足下述纳入标准和排除标准:1)纳入标准:2)样品合格性基于临床参数,包括下述受试者、结节和临床分期参数:a)受试者i)年龄≥40ii)任何吸烟状况,例如当前、以前或从未iii)共病状况,例如copdiv)处于临床缓解中最少5年的先前恶性肿瘤v)皮肤癌的既往史–鳞状或基底细胞b)结节i)放射学(1)大小≥4mm和≤70mm(直到2b期是合格的)(2)任何毛刺或磨玻璃影ii)病理学(1)恶性–腺癌、鳞状或大细胞(2)良性–炎性(例如肉芽肿、感染性)或非炎性(例如错构瘤)c)临床阶段i)原发性肿瘤:≤t2(例如1a、1b、2a和2b)ii)局部淋巴结:仅n0或n1iii)远处转移:仅m03)排除标准a)受试者:ipn诊断5年内的先前恶性肿瘤b)结节:i)大小数据不可用ii)对于癌症或良性spn,病理学数据不可用iii)病理学–小细胞肺癌c)临床阶段i)原发性肿瘤:≥t3ii)局部淋巴结:≥n2iii)远处转移:≥m1用于发现研究的效力分析发现研究的功效分析基于下述假设:1)总假阳性率()设定为0.05。2)用于多重检验的šidák校正用于计算用于测试200种蛋白质的有效,即,。3)有效样本大小降低0.864倍,以解释对于曼怀二氏检验比t检验更大的样品需求。4)基于先前经验,总变异系数设定为0.43。5)研究的效力(1-β)使用有效的和有效的样本大小,基于用于两样品、双侧t检验的公式进行计算。关于发现研究的效力通过样本大小/组群,以及对照样品和疾病样品之间的可检测倍数差异在表24中进行制表。表24:以给定概率检测蛋白质倍数变化所需的组群大小。用于验证研究的效力分析在验证研究中需要足够的癌症样品和良性样品,以确认从发现研究获得的排除分类器的性能。我们感兴趣的是对于排除分类器获得关于npv和ror的95%置信区间(ci)。使用本文的决策阈值选择节段中的等式,可以根据npv和ror衍生的灵敏度和特异性,即其中是预期用途群体中的癌症流行率。假设验证研究含有个癌症样品和个良性样品。基于二项式分布,灵敏度和特异性的方差通过以下给出使用本文的决策阈值选择节段中的等式,npv和ror的相应方差可以在大样品、正态分布近似下衍生为:。npv和ror的两侧95%ci分别由和给出,其中是正态分布的97.5%分位数。关于验证研究的预计95%ci通过样本大小/组群在表25中进行制表。肽和蛋白质测定的q值的计算为了确定假阳性测定率,如下计算肽srm测定的q值。使用来自不同蛋白质的跃迁之间的皮尔森相关分布作为零分布(图7),将经验性p值分配给在至少五个常见样品中检测到的来自同一肽的一对跃迁,否则分配‘na’的值。使用bioconductor中的“qvalue”软件包,将经验性p值转变为q值。对于表6中呈现的所有srm测定,肽q值都低于0.05。以相同的方式计算蛋白质srm测定的q值,除了将个别蛋白质的皮尔森相关性计算为来自蛋白质的不同肽的两个跃迁之间的相关性之外。对于在五个或更多个常见样品中未检测到两种肽的蛋白质,其q值无法正确地评估且分配‘na’。范畴混杂因素的影响表26.范畴混杂因素对分类器评分的影响。连续混杂因素的影响表27.连续混杂因素对分类器评分的影响。实施例8:用于肺结节的分子表征的系统生物学衍生的、基于血液的蛋白质组学分类器概述每年通过计算机断层摄影术发现了数百万个肺结节,但仍未被诊断为恶性或良性的。由于这些结节中的大多数是良性的,因此许多患者经历了不必要且昂贵的侵入性程序。本发明提出了用于鉴定良性结节的13蛋白质基于血液的分类器。使用系统生物学策略,鉴定了371种蛋白质候选物,并且对于每种蛋白质候选物开发了选择性反应监测(srm)测定。srm测定应用于多场所发现研究(n=143)中,其中良性和癌性血浆样品在结节大小、年龄、性别和临床场所方面进行匹配。13蛋白质分类器不是鉴定最佳性能的个别蛋白质,而是由在实验对象组上性能最佳的蛋白质形成。分类器对血浆样品的独立集合(n=104)进行验证,证实高阴性预测值(92%)和特异性(27%),其足够高以使四分之一的良性结节患者取消侵入性程序。重要的是,对非发现临床场所的验证性能显示了100%的npv和28%的特异性,支持了分类器的一般有效性。途径分析证实,分类器蛋白质很可能受几种转录调节剂(nf2l2、ahr、myc、fos)调节,所述转录调节剂与肺癌、肺部炎症和氧化性应激网络高度相关。值得注意的是,分类器评分不依赖于患者结节大小、吸烟史和年龄。由于这些是目前用于肺结节的临床管理的危险因素,因此这种分子测试的应用对于医生提供了强大的补充工具,以在肺癌诊断中使用。基本原理计算机断层摄影术(ct)每年鉴定了数百万个肺结节,其中许多未被诊断为恶性或良性的。这些结节中的绝大多数是良性的,但由于癌症的威胁,大量的良性结节患者经历了不必要的侵入性医疗程序,每年花费医疗保健系统数十亿美元。因而,存在关于非侵入性临床测试的未满足的迫切需要,所述临床测试可以以高概率鉴定良性结节。提出了用于鉴定良性结节的13蛋白质血浆测试或分类器。为了发现分类器,采取了基于以下推测的系统生物学方法:肿瘤中的生物学网络变得疾病扰乱的,并且改变其同源蛋白质的表达。这种系统方法采用各种策略来鉴定直接反映肺癌扰乱网络的血液蛋白质。首先,区分优先次序用于包括在分类器上的候选生物标记物是与正常肺细胞相比,由肺癌细胞的细胞表面分泌或脱落的蛋白质。这些既是与肺癌有关的蛋白质,也是最有可能由恶性肺结节释放到血液内的蛋白质。还调查了文献,以鉴定与肺癌有关的血液蛋白质。总共,从这三个来源衍生了用于包括在分类器上的388种蛋白质候选物的初始列表。另一种系统驱动的方法是通过388种蛋白质候选物如何频繁地出现在高性能蛋白质实验对象组上,将其区分优先次序用于包括在分类器上,与其个别诊断性能形成对照。这种策略的动机在于捕获肺癌扰动网络中蛋白质的整合行为的意图。频繁地出现在高性能实验对象组上的蛋白质称为协同蛋白。这是分类器的发现中的决定性步骤,因为大多数协同蛋白经常不是具有最佳个体性能的蛋白质。第三,根据其与肺癌网络的关系来解构分类器。理想地,分类器由来自多重肺癌扰动网络的多重蛋白质组成。我们猜想测量来自同一肺癌相关途径的多重蛋白质增加信噪比,因此增强分类器的性能。利用选择性反应监测(srm)质谱法(ms)来测量血浆中的候选蛋白质浓度。srm是ms的一种形式,其监测靶向蛋白的特定信息性(蛋白型或蛋白质特异性)肽的预定和高度特异性质量产物,称为跃迁。简言之,用于蛋白质的srm测定基于肽电离的高度再现性,ms的基础。在srm分析过程中,对质谱仪进行编程,以监测待测定的特定蛋白质的跃迁。将所得到的层析图整合,以提供定量或半定量的蛋白质丰度信息。srm测定的益处包括高蛋白特异性,大型复用能力,以及快速可靠的测定开发和部署。srm多年来一直用于小分子分析物的临床测试,并且最近还用于生物学相关测定的开发。存在特别的公共资源,以加速srm测定开发,包括peptideatlas、plasmaproteomeproject、srmatlas和peptideatlassrmexperimentallibrary。根据用于临床测试开发的不断发展的指导,使用来自多重临床场所的独立血浆集合发现分类器(n=143),并且进行验证(n=104),所述独立血浆集合与具有肺部结节的患者的预期用途群体一致,所述肺部结节定义为大小至多30mm的圆形不透明物。与其它生物标记物研究(其利用与广泛的肺癌临床谱(i至iv期)有关的生物样本)形成对比,分析的癌症血浆样品仅限于ia期,其对应于大小30mm或更小的肺部结节的预期用途群体。如由临床医生的诊断需要指导的,分类器通过参数例如年龄、吸烟史或结节大小,产生可对预期用途的进一步临床分层修正的性能。13蛋白质分类器的验证性能证实了92%的阴性预测值(npv)和27%的特异性。为了临床实用性,分类器必须可靠地且频繁地提供可以参与医生决定的信息,以避免侵入性程序。要求高npv,以确保分类器可靠地鉴定良性结节。同样地,恶性结节很少(8%或更少)由分类器报告为良性的。27%的特异性暗示四分之一的良性结节患者可以避免侵入性程序,并且因此,频繁地提供临床实用性信息。所有验证样品都不依赖于发现样品,并且37个来自新的临床场所。对来自新场所的样品的性能证实100%的npv和28%的特异性,提示分类器性能扩展到新的临床环境。值得注意的是,分类器评分被证实为不依赖于患者的年龄、吸烟史和结节大小,从而以用于评估肺结节的疾病状态的信息分子尺度补充了当前的临床危险因素。结果表28呈现了在最初的388种蛋白质候选物改进为用于验证和性能评价的13种分类器蛋白质的集合中采取的步骤。结果以相同顺序呈现。表28.在388种候选物改进为13蛋白质分类器中的步骤用于测定开发的生物标记物候选物选择。为了鉴定血液中从肺肿瘤细胞脱落或分泌的肺癌生物标记物,使用与质谱法组合的细胞器分离技术,从新鲜切除的肺肿瘤中鉴定了相对于正常肺细胞,在细胞表面上过表达或由肺癌肿瘤细胞过度分泌的蛋白质。另外,使用公共和私人资源,执行了关于肺癌生物标记物的广泛文献检索。组织来源的生物标记物和文献来源的生物标记物均要求具有在血液中先前检测到的证据。组织(217)和文献(319)候选物重叠了148种蛋白质,导致388种蛋白质候选物的列表。srm测定的开发。标准的合成肽技术用于由388种蛋白质候选物开发371蛋白质复用srm测定。对于17种候选物,无法开发或可靠地鉴定适当的合成肽。将371个srm测定应用于来自患者的血浆样品,所述患者具有病理学确认的良性结节和病理学确认的恶性肺部结节,以确定在血浆中可以检测到371种蛋白质中的多少种。总共190个srm测定能够检测其在血浆中的靶蛋白(51%成功率)。这一成功率(51%)与类似努力(16%)非常有利地比较,以开发大规模srm测定用于检测血液中的多样化癌症标记物。在血液中检测到的190种蛋白质中,114种衍生自组织来源的候选物,而167种衍生自文献来源的候选物(91种蛋白质重叠)。猜想血液中未检测到49%的候选蛋白质是存在的,但低于该技术的检测水平。分类器发现。表29中显示了用于分类器发现的143个样品的特点概括。从三个临床场所获得样品,以避免对单个临床场所的过度拟合。参与的临床场所是institutuniversitairedecardiologieetdepneumologiedequebec(iucpq)、newyorkuniversity(nyu)和universityofpennsylvania(upenn)。所有样品都选择为与预期用途一致,具体地,具有30mm或更小的结节大小。癌症样品和良性样品得到病理学确认。表29.发现研究和验证研究中的受试者和结节的临床特征*显示的数据是具有括号中指示的四分位数范围的中值。†曼怀二氏检验。‡费氏精确测试。§从不吸烟者定义为一生中吸烟少于100支香烟的个体。¶包年定义为吸烟总年数与每天吸烟的平均包数的乘积。对于发现集中的4个癌症受试者和6个良性受试者、以及验证集中的2个癌症受试者和3个良性受试者,包年数据不可用。||iucpq是instituteuniversitairedecardiologieetdepneumologiedequebec。**对于发现研究,良性诊断的“其它”范畴包括:淀粉样变性,n=2;纤维弹性结节,n=1;纤维化,n=1;出血性梗塞,n=1;淋巴样聚集体,n=1;机化性肺炎,n=3;肺梗塞,n=1;硬化性血管瘤,n=1;以及胸膜下纤维化伴良性淋巴样增生,n=1。对于验证研究,良性诊断的“其它”范畴包括:淀粉样变性,n=1;支气管上皮细胞,n=4;细支气管炎间质纤维化,n=1;肺气肿,n=1;纤维化炎性病变,n=1;炎症,n=1;实质性肠套叠,n=1;淋巴管瘤,n=1;混合淋巴细胞和组织细胞,n=1;正常实质,n=1;机化性肺炎,n=1;肺梗塞,n=2;呼吸性细支气管炎,n=1;以及鳞状化生,n=1。††对于发现研究,非小细胞肺癌(nsclc)诊断“其它”范畴包括:腺癌鳞状细胞混合型,n=1;大细胞鳞状细胞混合型,n=1;多形性癌,n=1,以及未指定,n=1。对于验证研究,nsclc诊断“其它”范畴包括:类癌,n=2;大细胞鳞状细胞混合型,n=1;以及未指定,n=2。通过在年龄、性别、结节大小和临床场所方面的匹配,来配对良性样品和癌症样品,以避免在srm分析期间的偏差,以及确保发现的生物标记物不是年龄、性别、结节大小或临床场所的标记物。将371-蛋白质srm测定应用于143个发现样品,并且使用逻辑回归模型分析所得到的跃迁数据,以衍生13蛋白质分类器(表30)。这种改进中的关键步骤(表28)是鉴定36种协同蛋白,其中21种具有稳固的srm信号。如果在最佳性能的实验对象组上比单独偶然预期的更频繁地发现,则蛋白质被视为协同的,其显著性使用下述统计估计程序确定。简言之,生成一百万个随机的10蛋白质实验对象组,并且计算每种蛋白质在最佳性能实验对象组中的频率(p值≤10-4)。这些蛋白质从在良性样品或癌症样品中可再现地检测到的125种蛋白质列表中取样(参见表28)。估计程序和完整发现过程的全部细节在实施例9的材料和方法中进行描述。重要的是,在执行验证前已完全定义了13蛋白质分类器。表30.13蛋白质逻辑回归分类器常数等于36.16。。分类器验证。总共52个癌症样品和52个良性样品(表29)用于验证13蛋白质分类器的性能。所有验证样品都来自与发现样品不同的患者。另外,36%的验证样品来源自新的第四个临床场所,vanderbiltuniversity(vanderbilt)。参与验证研究的新临床场所提供了分类器的性能泛化超出发现研究的更大信心。剩余的验证样品是从发现场所中随机选择的。样品选择为与预期用途一致,并且如发现研究是匹配的。将分类器应用于验证样品并进行分析(实施例9中的材料和方法)。分类器的性能根据阴性预测值(npv)和特异性(spc)呈现于图12中,因为这是两个最临床相关的测量。npv是由分类器预测为良性结节是真正良性的基于群体的概率。由于npv代表了分类器对预期用途群体的性能,因此它可以根据分类器的灵敏度、特异性和预期用途群体中的估计癌症流行率(20%)来计算。特异性是由分类器预测为良性的良性结节的百分比。分类器生成范围为0至1的癌症概率评分。可以这样限定这个范围内的任何参考值,使得如果样品的分类器评分低于参考值,则样品被预测为良性的,或者如果样品的分类器评分高于参考值,则被预测为恶性的。在实践中使用的参考值主要取决于医生及其最低要求的npv。为了说明的目的,我们假设npv要求为90%。在参考值0.43下,分类器具有对于发现样品96%+/-4%的npv以及45%+/-13%的特异性,其中报告了95%的置信区间。在相同的参考值0.43下,分类器具有对于验证样品92%+/-7%的npv和27%+/-12%的特异性。表31报告了分类器对于发现和验证样品集以及多重肺癌流行率的性能。对于每种肺癌流行率,参考值选择为确保npv为90%或更高。表31.分类器在三种癌症流行率下在发现和验证中的性能npv为阴性预测值。ppv为阳性预测值。13蛋白质分类器对来自新临床场所(范德比尔特)的验证样品的性能是该分类器对未来样品的性能的重要指标,以及该分类器不会对三个发现场所过度拟合的强烈标志。在相同的参考值0.43下,关于vanderbilt样品的npv和特异性分别为100%和28%。图13呈现了分类器对所有247个发现和验证样品的应用。图13将吸烟(以包年测量)和结节大小(与每个圆圈的直径成比例)的临床危险因素与分配给每个样品的分类器评分进行比较。结节大小似乎没有随着分类器评分而增加。实际上,大结节和小结节两者均在分类器评分谱中分散。为了定量这一观察,计算了分类器评分与结节大小、吸烟史包年和年龄之间的皮尔森相关性,并且发现是无关紧要的(表32)。这一观察的含义是显著的。分类器提供了关于肺结节的疾病状况的信息,其不依赖于当前使用的关于恶性肿瘤的三种风险因素(年龄、吸烟史和结节大小),并且因此提供了具有极大附加临床价值的增量分子信息。关于结节大小相对于分类器评分的类似图,参见图15。表32.临床特征对分类器评分的影响分类器的分子基础。为了解决这13种分类器蛋白质的生物学相关性,将它们提交,用于使用ipa(ingenuitysystems,www.ingenuity.com)的途径分析。鉴定最可能引起这13种蛋白质的调节的转录因子。使用标准ipa分析参数,四种最重要的(参见实施例9中的材料和方法)核转录调节剂是fos(原癌基因c-fos)、nf2l2(核因子红系2相关因子2)、ahr(芳烃受体)和myc(myc原癌基因蛋白)。这些蛋白质调节13种分类器蛋白质中的12种,其中islr是例外(参见下文)。fos是许多形式的癌症所共有的。nf2l2和ahr与肺癌、氧化性应激应答和肺部炎症有关。myc与肺癌和氧化性应激应答有关。这四种转录调节因子和13种分类器蛋白质共同地也与相同的三个生物学网络(即,肺癌、肺部炎症和氧化性应激应答)高度相关(p值1.0e-07)。这概括于图14中,其中描绘了分类器蛋白(绿色)、转录调节剂(蓝色)和三个合并的网络(橙色)。尽管只有islr(含有富含亮氨酸重复蛋白的免疫球蛋白超家族)没有通过这三个网络与其它分类器蛋白质连接,但它通过对肺非特异性的癌症网络连接。总之,13种分类器蛋白的调节可以向后连接到与肺癌、肺部炎症和氧化性应激应答网络高度相关的几种转录调节剂;反映肺癌方面的三个生物学过程。本发明以多重方式脱颖而出。首先,跨越多重流行率估计值,13蛋白质分类器的性能达到在验证中至少90%或更高的npv(和灵敏度)的预期用途性能要求(参见表31)。其次,与其中使用范围为i期至iv期的非预期用途样品的先前研究形成对比,在发现和验证使用了预期用途群体样品(结节大小30mm或更小和/或ia期)。在一些情况下,先前的工作中并未公开结节大小信息。第三,13蛋白质分类器证实为提供不依赖于当前使用的结节大小、吸烟史和年龄的癌症风险参数的评分。与如复用抗体的技术(其对于复用关于特异性疾病的数百种候选标记物经常不可行)形成对比,srm技术的利用使得能够全面询问与肺癌过程相关的蛋白质。临床研究设计。生物标记物研究的设计和进行必然受到临床试验的最终预期用途群体和性能要求的影响。新出现的指导帮助设计具有更大机会转化为临床影响的研究。在此处呈现的发现研究和验证研究的设计中,四个要求是尤其重要的。首先,进行多重临床场所发现研究使我们能够确定对变化稳固的那些蛋白质,所述变化通过场所间的样品加工和管理中的差异引入,以及来自由不同场所医院所服务的群体中的任何生物学差异。此类设计是至关重要的,因为场所间的变化来源经常可以超过生物信号。其次,在发现和验证阶段利用如通过年龄、吸烟史和结节大小限定的预期用途样品,使我们能够获得分类器的性能范围的真实估计值。第三,癌症组群和良性组群在年龄、性别、结节大小和临床场所方面的仔细匹配,不仅在避免偏差中是至关重要的,而且在分类器的发现和验证中也是至关重要的,所述分类器提供了不依赖于这些临床因素以及吸烟史的评分。第四,验证样品来自与发现样品不同的患者。此外,36%的验证样品来自全新的临床场所,显示结果并未对发现阶段中使用的场所过度拟合的关键验证步骤。对来自新临床场所的样品的性能格外地高(100%的npv和28%的特异性),产生了测试在临床实践中的性能的高可信度。系统生物学和血液标记。用srm技术将系统生物学方法与生物标记物发现整合,允许同时探索大量肺癌相关的蛋白质,导致高度灵敏的分类器。系统方法采用了几种策略。首先,鉴定从肺癌细胞的细胞表面分泌或脱落的蛋白质(即,组织来源的),因为这些很可能是待在血液中检测的肺癌扰乱的蛋白质。在分类器的13种蛋白质中,7种是组织来源的,证实组织来源是用于将蛋白质区分优先次序用于srm测定开发的有效方法。第二种系统驱动的方法是最协同的蛋白质生物标记物的鉴定。协同蛋白是可能并非最佳的个别表现者,但频繁地出现在高性能实验对象组中的那些蛋白质。这种方法的动机是衍生具有来自多重肺癌相关网络的多重蛋白质的分类器的愿望。通过监测多重蛋白质和网络,预期分类器对恶性结节的循环标记是高度敏感的,如验证中证实的。存在协同蛋白方法的有效性的两种确认。途径分析证实,分类器蛋白质很可能受少数转录调节剂(ahr、nf2l2、myc、fos)调节,所述转录调节剂与肺癌、肺部炎症和氧化性应激应答网络/过程高度相关。慢性肺部炎症和氧化性应激应答两者均与nsclc的发展联系。分类器的优势在于它监测来自这些多重肺癌相关过程的多重蛋白质。这种多重蛋白质、多重过程调查解释了分类器用于检测由恶性结节发出的循环标记的高灵敏度,以及因此,当分类器将结节称为良性时的高npv。协同方法的第二种验证是与传统生物标记物策略的直接比较。通常,通过关于个别诊断性能过滤,蛋白质在发现过程中入围。为了对比与在高性能实验对象组上的频率相对的基于强个体性能来过滤蛋白质之间的差异,我们使用曼怀二氏非参数检验计算了关于每种蛋白质的p值。36种协同蛋白中只有2种具有低于0.05的p值,对于测量个体性能的常用显著性阈值。更重要的是,我们使用用于13蛋白质分类器衍生的相同步骤(参见表28以及实施例9中的材料和方法)来衍生“p分类器”,除了使用曼怀二氏p值代替协同评分之外。与13蛋白质分类器在发现中的npv96%和特异性45%、以及在验证中的npv92%和特异性27%相比,p分类器实现了在发现中的npv96%和特异性18%、以及在验证中的npv91%和特异性19%。注意,参考值阈值选择为确保至少90%的npv。因此,我们预期13蛋白协同分类器和p分类器之间相似的高npv性能。特异性是其中可以进行比较的性能测量。这是观察到从13蛋白协同分类器到p分类器的性能中的显著下降的地方。这确认了最佳的个别蛋白质表现者不一定是用于分类器的最佳蛋白质。最具信息性的蛋白质。分类器中哪些蛋白质是最具信息性的为了回答这个问题,所有可能的分类器都由稳固的协同蛋白集合构建,并且测量其性能。确定了100个最佳性能的实验对象组中每种蛋白质的频率。四种蛋白质(lrp1、coia1、aldoa、lg3bp)是高度富集的,其中100个最佳分类器中的95%具有这四种蛋白质中的至少三种(p值<1.0e-100)。八种蛋白质中的七种(lrp1、coia1、aldoa、lg3bp、bgh3.prdx1、tetn、islr)一起出现在超过一半的所有最佳分类器中(p值<1.0e-100)。注意到13蛋白质分类器含有另外的蛋白质,因为它们进一步增加性能,可能是通过测量相同的三个肺癌网络(肺癌、肺部炎症和氧化性应激)中的蛋白质。结论是用于肺结节表征的协同蛋白的高性能实验对象组在组成上彼此相似,伴随对于特别信息性的(协同)蛋白集合的优先。总之,通过整合用于生物标记物发现(具有癌症相关性的组织来源的候选物、协同蛋白、来自多重肺癌相关网络的多重蛋白质)的系统生物学策略、使能技术(用于总体蛋白质组学查询的srm)和临床重点(设计研究用于预定用途),本发明鉴定了13蛋白质蛋白质组学分类器,其提供了有关肺结节的疾病状态的分子了解。实施例9:材料和方法候选血浆蛋白质的鉴定。两种方法用于鉴定用于肺癌分类器的候选蛋白质,包括具有nsclc的组织病理学诊断的肺组织的蛋白质组学分析、以及对于肺癌相关蛋白质的文献数据库检索。还评价了所有候选蛋白质的血液循环证据,并且满足了一种或多种证据要求。使用srm-ms分析血浆样品。简言之,用于血浆等分试样的srm-ms分析的方案包括中和高丰度蛋白质在igy14-supermix树脂柱(sigma)上的免疫耗竭,变性,胰蛋白酶消化和脱盐,随后为反相液相层析和所得肽样品的srm-ms分析。srm测定的开发。如先前所述,基于合成肽开发了用于候选蛋白质的srm测定。在至多五种合适的肽/蛋白质的鉴定和合成后,对于双重荷电和三重荷电的前体离子,在5500qtrap®质谱仪上收集srm触发的ms/ms波谱。使用mascot以及15的最低截止评分,将获得的ms/ms波谱分配给个别肽。然后选择至多四个跃迁/前体离子用于优化。所得到的相应的最佳保留时间、去簇电压和碰撞能对于所有跃迁进行组装。对所有合成肽的混合物以及两个合并的血浆样品测量了最佳跃迁,所述血浆样品各自得自在institutuniversitairedecardiologieetdepneumologiedequebec(iucpq,quebec,canada),具有良性或恶性(即nsclc)肺部结节的十个受试者。在由该机构的伦理审查委员会(erb)批准的研究中,所有受试者都提供了知情同意书并贡献了生物样本。如上所述加工血浆样品。通过srm-ms分析了1750个跃迁的分批,其中人工审查srm数据,以选择两种最佳的肽/蛋白质和两个最佳的跃迁/肽。定义为合成肽混合物中肽的两个最佳跃迁的强度之间的比率的强度比,用于评价生物样品中跃迁的特异性。不选择证实由其它跃迁干扰的跃迁。开发了确保观察到的跃迁对应于它们预期测量的肽和蛋白质的方法。特别是,开发的93%的肽跃迁具有低于5%的错误率。发现研究设计。使用先前得自受试者的存档的k2-edta血浆等分试样,执行回顾性、多中心、病例对照研究,在分别由在iucpq或newyorkuniversity(newyork,ny)和universityofpennsylvania(philadelphia,pa)的伦理审查委员会(erb)或机构审查委员会(irb)批准的研究中,所述受试者提供了知情同意书并贡献了生物样本。另外,根据需要由分别机构的erb或irb审查并批准申办者的研究方案后,由研究人员提供血浆样品。用于蛋白质组学分析的样品合格性基于研究纳入标准和排除标准的满足,包括受试者的人口统计学信息、受试者通过胸部ct扫描和30mm的最大线性尺度的相应肺部结节射线照相特征、以及在诊断性手术切除时获得的肺部结节的组织病理学,即nsclc或良性(即非恶性)过程。在合格的样品中,每个癌症良性样品对尽可能按性别、结节大小(±10mm)、年龄(±10岁)、吸烟史包年(±20包年)以及中心进行匹配。为了确保临床数据的准确度,根据由1996年的健康保险携带和责任法案(hipaa)制定的指导,执行与受试者和肺部结节两者相关的临床数据的独立监测和验证,以确保受试者隐私权。该研究具有92%的概率检测恶性和良性肺部结节之间的蛋白质丰度中的1.5倍差异。逻辑回归模型。逻辑回归分类方法用于将跃迁实验对象组组合到分类器内,并且对于每个样品计算在0和1之间的分类概率评分。样品的概率评分()确定为,(1)其中是样品s中对数转换(底数2)的标准化的跃迁强度,是相应的对数回归系数,是分类器特异性常数,并且是分类器中的跃迁总数目。如果小于参考值,则样品被分类为良性的,否则为癌症。参考值可以根据所需的npv来增加或减少。为了定义分类器,必须从发现研究学习(即训练)跃迁(即蛋白质)实验对象组、其系数、标准化跃迁、分类器系数和参考值,然后使用验证研究进行确认。肺部结节分类器的开发。发现研究的目标是衍生对于在预期用途群体中的临床实用性具有足够靶性能的多变量分类器,即,具有90%或更高的npv的分类器。这一目标已掺入数据分析策略中。分类器开发包括下述:原始srm-ms数据的标准化和过滤;鉴定在顶部性能的实验对象组中以高频率出现的候选蛋白质;基于srm-ms信号质量评估候选蛋白质;基于其在性能中的稳定性,选择候选蛋白质用于最终分类器;并且对逻辑回归模型训练,以衍生最终分类器。表28提供了主要步骤的概括性概述。使用六种内源蛋白质的实验对象组,执行原始srm-ms数据的标准化,以降低样品间的强度变化。在数据标准化后,将srm-ms数据过滤到这样的跃迁,其具有相应蛋白质的最高强度,并且满足用于在最少50%癌症或50%良性样品中的检测的标准。总共125种蛋白质满足了可再现检测的这些标准。在所有样品中,缺失值被替换为相应跃迁的最小检测值的一半。剩余的跃迁然后用于鉴定定义为协同蛋白的蛋白质,其在顶部性能的蛋白质实验对象组中以高频率出现。协同蛋白使用下述估计程序衍生,因为评估所有可能的蛋白质实验对象组的性能在计算上是不可行的。对1x106个实验对象组执行蒙特卡洛交叉验证(mccv)(36),每个实验对象组包含10种随机选择的蛋白质,并且如上所述,使用20%留出率和102个样品排列拟合至逻辑回归模型。生成每个实验对象组的接受者操作特征(roc)曲线,并且在roc曲线(auc)下,但在90%灵敏度的边界上方的相应部分的面积(定义为部分auc(37、38)),用于评价实验对象组的性能。通过集中于各个实验对象组在高灵敏度区域的性能,部分auc允许鉴定在npv上具有高且可靠的性能的实验对象组。以大于偶然预期的频率出现在顶部100个性能实验对象组中的候选蛋白质被鉴定为协同蛋白。对于每种蛋白质,协同评分定义为其在100个高性能实验对象组上的频率除以预期频率。高度协同蛋白具有1.75或更高的评分(相应的单侧p值<0.05),而非协同蛋白具有1或更低的评分。注意,取样一百万个实验对象组,以确保100个顶部性能的实验对象组是出色的(经验p值≤10-4)。另外,基于较大的实验对象组并不改变所得到的协同蛋白列表的经验证据,在该程序中使用了大小为10的实验对象组。我们还希望避免对逻辑回归模型的过度拟合。总共鉴定了36种协同蛋白,包括15种高度协同蛋白。然后,人工检查了协同蛋白的所有跃迁的原始层析图。具有低信噪比和/或显示任何干扰的迹象的蛋白质已从用于最终分类器的进一步考虑中去除。总共鉴定了21种协同且稳固的蛋白质。然后以迭代、逐步的程序评估剩余的候选蛋白质,以衍生最终分类器。在每个步骤中,使用20%的留出率和104个样品排列执行mccv,以对逻辑回归模型训练剩余的候选蛋白质,并且评价通过模型对于每种蛋白质衍生的系数的变化性,即稳定性。鉴定并去除具有最不稳定的系数的蛋白质。当相应的部分auc最佳时,鉴定用于最终分类器的蛋白质。最终分类器中的13种蛋白质中7种是高度协同的。通过mccv以20%的留出率和2x104个样品排列,将最终分类器中的蛋白质进一步对逻辑回归模型进行训练。肺部结节分类器验证。验证研究的设计与发现研究的设计等同,但涉及与发现研究中未评估的独立受试者和独立肺部结节相关的k2-edta血浆样品。另外的样本得自vanderbiltuniversity(nashville,tn),具有对于患者同意书、irb批准和hipaa要求的满足的相似要求。在验证研究中的104种癌症样品和良性样品中,一半在发现研究后立即进行分析,而另一半在以后进行分析。操纵该研究以观察到90±8%的npv的预期95%置信区间(ci)。验证研究中的原始srm-ms数据集以与发现数据集相同的方式标准化。通过利用两项研究中的人血浆标准(hps)样品作为外部校准物,可以减轻发现研究与验证研究之间的变化性。然后,将验证研究中的缺失数据替换为发现研究中的相应跃迁的最小检测值的一半。将跃迁强度应用于先前在训练阶段学习的最终分类器的逻辑回归模型,由其将分类器评分分配给各个样品。然后基于分类器评分评价肺部结节分类器对验证样品的性能。ipa途径分析。使用标准参数。具体地,在核转录调节剂的搜索中,要求为p值<0.01,伴随调节的最小3种蛋白质。使用ipa知识数据库作为背景,使用右尾费氏精确检验来确定显著性。候选生物标记物鉴定。通过组织蛋白质组学鉴定的候选生物标记物。在同一肺叶中切除的nsclc(腺癌、鳞状细胞和大细胞)肺肿瘤和非相邻的正常组织的样本得自患者,所述患者在通过在centrehospitalierdel’universitédemontréal和mcgilluniversityhealthcentre的伦理审查委员会批准的研究中提供了知情同意书。肺肿瘤组织的蛋白质组学分析靶向内皮细胞(腺癌,n=13;鳞状细胞,n=18;以及大细胞,n=7)和上皮细胞(腺癌,n=19;鳞状细胞,n=6;以及大细胞,n=5)上的膜结合蛋白,以及与高尔基体有关的蛋白质(腺癌,n=13;鳞状细胞,n=15;以及大细胞,n=5)。在缓冲液中洗涤并用polytron破坏以制备匀浆物后,从来自新鲜肺切除物的正常组织或肿瘤组织中分离来自内皮细胞或上皮细胞的膜蛋白和分泌性蛋白。细胞膜方案包括将上清液使用180µm筛孔过滤,以及在4°c下以900xg离心10分钟,然后在50%(w:v)蔗糖上分层,以及在4°c下以218,000xg离心1小时,以使膜形成团块。将膜团块重悬浮并用微球菌核酸酶处理,并且与通过质膜类型指定的下述抗体一起温育:内皮膜(抗血栓调节蛋白、抗ace、抗cd34和抗cd144抗体);上皮膜(抗esa、抗cea、抗cd66c和抗ema抗体),然后在4°c下以280,000xg(内皮)或218,000xg(上皮)在50%(w:v)蔗糖垫的顶部上离心1小时。在团块重悬浮后,使用macs微珠分离质膜。用ki处理内皮细胞质膜,以去除细胞质外周蛋白。上皮细胞质膜的洗脱物在4°c下以33,000xg在33%(w:v)蔗糖垫上离心30分钟,伴随在上清液和蔗糖垫去除后,将团块重悬浮于laemmli/尿素/dtt中。为了分离分泌性组织蛋白,将组织匀浆物(如上所述制备)的密度调整至1.4m蔗糖,然后通过在0.8和1.2m的不连续蔗糖梯度上,在4°c下以100,000xg等密度离心2小时来分离分泌囊泡。收集在0.8/1.2m界面处浓缩的囊泡,并且与0.5mkcl进一步温育25分钟,以去除松散结合的外周蛋白。通过在4°c下以150,000xg超速离心1小时来回收囊泡,然后在4°c下用100mm(nh4)hco3(ph11.0)打开30分钟。在4°c下以150,000xg超速离心1小时之后,从上清液中回收分泌性蛋白。然后通过cellcarta®(caprion,montréal,québec)蛋白质组学平台分析膜蛋白或分泌性蛋白,包括通过胰蛋白酶消化,通过强阳离子交换层析法分离,以及通过与电喷雾串联质谱法(ms/ms)偶联的反相液相层析分析。样品中的肽通过使用mascot的ms/ms波谱的数据库检索进行鉴定,并且基于其在样品中的信号强度通过无标记方法进行定量,类似于文献中所述的那些。然后将其肿瘤/正常丰度比≥1.5或≤2/3的蛋白质鉴定为候选生物标记物。通过文献检索鉴定的候选生物标记物。使用预定义术语和自动化perl脚本,对下述数据库执行自动化文献检索:2010年5月6日uniprot,2010年5月17日entrez、nbk3836,以及2010年7月8日nextbio。候选生物标记物被编译并使用uniprotknowledgebase映射到uniprot标识符。血液中候选生物标记物的存在。组织和文献鉴定的生物标记物候选物被要求证实作为可溶性或增溶的循环蛋白在文献或数据库中的记录证据。第一个标准是通过质谱法检测加以证明,其中候选物如先前通过下述数据库特异性标准进行指定:hupo9504中最少2种肽,所述hupo9504含有通过ms/ms鉴定的9,504种人蛋白;hupo889中最少1种肽,所述hupo889是含有889种人蛋白的hupo9504的较高置信度子集;或peptideatlas(2009年11月构建)中的至少2种肽。第二个标准是在uniprot中注释为分泌性蛋白或单次跨膜蛋白。第三个标准在文献中被指定为血浆蛋白质。第四个标准是基于各种程序的使用预测为分泌性蛋白:通过tmhmm预测为具有一个跨膜结构域的蛋白质,然而,其基于通过signalp的预测是切割的;或通过tmhmm预测为不具有跨膜结构域,以及通过signalp或secretomep预测为分泌性蛋白。所有候选蛋白质都满足一个或多个标准。研究设计和效力分析。样品、受试者和肺部结节的纳入和排除标准。关于血浆样品的纳入标准是在含edta的血液管中收集;得自在参与机构先前入选伦理审查委员会(erb)或机构审查委员会(irb)批准的研究的受试者;并且存档的,例如如通过研究方案要求的标记、等分并冷冻的。关于受试者的纳入标准如下:年龄≥40;任何吸烟状况,例如当前、以前或从不;任何共病状况,例如慢性阻塞性肺疾病(copd);处于临床缓解中最少5年的任何先前恶性肿瘤;任何皮肤癌的既往史,例如鳞状或基底细胞。唯一的排除标准是肺部结节诊断5年内的先前恶性肿瘤。关于肺部结节的纳入标准包括放射学、组织病理学和分期参数。放射学标准包括大小≥4mm和≤30mm,以及任何毛刺或磨玻璃影。组织病理学标准包括恶性肿瘤,例如非小细胞肺癌(nsclc),包括腺癌(和细支气管肺泡癌(bac)、鳞状或大细胞),或良性过程,包括炎性(例如肉芽肿、感染性)或非炎性(例如错构瘤)过程的诊断。临床分期参数包括:原发性肿瘤:≤t1(例如1a和1b);局部淋巴结:仅n0或n1;远处转移:仅m0。关于肺部结节的排除标准包括下述:结节大小数据不可用;小细胞肺癌的组织病理学诊断的病理学数据不可用;以及下述临床分期参数:原发性肿瘤:≥t2,局部淋巴结:≥n2,以及远处转移:≥m1。样品布局。至多15个配对样品/分批迭代随机且迭代地分配给实验加工分批,直到在年龄、性别或结节大小方面无法证实统计偏差。进一步将每个加工分批内的配对样品随机且重复地分配至加工分批内的位置,直到位置与年龄、性别和结节大小之间的相应皮尔森相关系数的绝对值小于0.1。随后将每对癌症样品和良性样品随机分配到其在分批中的相对位置。为了提供用于质量评价的阳性对照,将合并的人血浆标准(hps)(bioreclamation,hicksville,ny)的三个200µl等分试样分别放置在每个加工分批的开始、中间和结尾。分批内的样品一起进行分析:在免疫耗竭和srm-ms分析中序贯地,但在变性、消化和脱盐期间平行地。用于分类器发现研究的效力分析。发现研究的功效分析基于下述假设:(a)总假阳性率()设定为0.05。(b)用于多重检验的šidák校正用于计算用于测试200种蛋白质的有效,即,。(c)有效样本大小降低0.864倍,以解释对于曼怀二氏检验比t检验更大的样品需求(13)。(d)基于先前经验,总变异系数设定为0.43。(e)研究的效力(1-β)使用有效的和有效的样本大小,基于用于两样品、双侧t检验的公式进行计算。用于分类器验证研究的效力分析。在验证研究中需要足够的癌症样品和良性样品,以确认从发现研究获得的肺部结节分类器的性能。我们感兴趣的是对于分类器获得关于npv和特异性的95%置信区间(ci)。假设肺部结节的癌症流行率为,如下根据灵敏度()和特异性()计算分类器对具有肺部结节的患者群体的阴性预测值(npv)和阳性预测值(ppv):(s1)(s2)使用上文的等式(s1),可以根据npv和特异性衍生灵敏度,即(s3)假设验证研究含有个癌症样品和个良性样品。基于二项式分布,灵敏度和特异性的方差通过以下给出:(s4)(s5)使用上文的等式(s1、s2),npv和ppv的相应方差可以在大样品、正态分布近似下衍生为:,(s6)。(s7)然后,灵敏度、特异性、npv和ppv的两侧95%ci分别由、、和给出,其中是正态分布的97.5%分位数。实验程序。免疫亲和层析。通过分别向玻璃层析柱(tricorn,gehealthcare)中添加10ml的含有2:1比率的igy14和supermix树脂(sigmaaldrich)的50%浆料来制备免疫亲和柱,并且允许通过重力沉降,在柱中产生5ml体积的树脂。将柱加盖并且放置在hplc系统(agilent1100系列)上,用于用0.15m(nh4)hco3以2ml/分钟进一步填充20分钟,其性能通过hps等分试样的重复注射进行评价。在每个样品分批的免疫亲和分离之前评价柱性能。为了分离低丰度蛋白质,将60µl血浆在0.15m(nh4)hco3(1:2v/v)中稀释至180µl最终体积,并且使用0.2µmacroprep96孔滤板(palllifesciences)进行过滤。使用3种缓冲液(上样/洗涤:0.15m(nh4)hco3;汽提/洗脱:0.1m甘氨酸,ph2.5;以及中和:0.01mtris-hcl和0.15mnacl,ph7.4),在连接到hplc系统(agilent1100系列)的igy14-supermix柱上进行免疫亲和分离,其循环包括上样、洗涤、洗脱、中和以及再平衡,持续36分钟。在280nm处监测未结合级分和结合级分,并且在分离后分辨基线。收集未结合级分(含有低丰度蛋白质),用于下游加工和分析,并且在酶促消化之前进行冻干。酶促消化和固相提取。在温和的变性条件下,在200μl1:1的0.1m(nh4)hco3/三氟乙醇(tfe)(v/v)中重构后,含有低丰度蛋白质的冻干级分用胰蛋白酶进行消化,然后允许在定轨振荡器上在rt下温育30分钟。将样品在800μl0.1m(nh4)hco3中稀释,并且用0.4µg胰蛋白酶(princetonseparations)/样品在37°c下消化16小时且冻干。将冻干的胰蛋白酶肽在350μl0.01m(nh4)hco3中重构,并且在定轨振荡器上在rt下温育15分钟,随后为使用30μl0.05mtcep的还原以及在rt下的1小时温育,以及在375µl90%水/10%乙腈/0.2%三氟乙酸中的稀释。提取板(emporec18,3mbioanalyticaltechnologies)根据制造商的方案进行条件化,并且在样品上样后,在500µl95%水/5%乙腈/0.1%三氟乙酸中洗涤,并且通过200µl52%水/48%乙腈/0.1%三氟乙酸洗脱到收集板内。将洗脱物拆分成2个相等的等分试样,并且在真空浓缩器中干燥。一个等分试样立即用于质谱法,而另一等分试样被贮存于-80°c下。紧在lc-srmms分析之前,在12μl90%水/10%乙腈/0.2%甲酸中重构样品。srm-ms分析。使用毛细管反相lc柱(thermobiobasic18kappa;柱尺寸:320μmx150mm;粒度:5μm;孔径:300å)和纳米hplc系统(nanoacquity,watersinc.),将肽样品分开。流动相为(a)0.2%甲酸的水溶液和(b)0.2%甲酸的乙腈溶液。将样品注入(8μl),并且使用线性梯度(98%a至70%a)以5μl/分钟分离19分钟。肽被直接洗脱到质谱仪(5500qtraplc/ms/ms,absciex)的电喷雾源内,所述质谱仪以预定的srm正离子模式运行(q1分辨率:单位;q3分辨率:单位;检测窗口:180秒;循环时间:1.5秒)。然后通过软件multiquant(absciex)整合跃迁强度。10,000的强度阈值用于过滤出非特异性数据和未检测到的跃迁。原始srm-ms数据的标准化和校准。耗竭柱漂移的定义。由于在每个免疫亲和柱的重复使用后观察到的信号强度中的改变,通过定量对照hps样品中的跃迁强度来评价柱的性能。假设是hps样品中的跃迁强度,样品的漂移定义为(s8)其中是由同一柱耗竭的所有hps样品中的平均值,并且中值取自样品中所有检测到的跃迁。柱的变化性或漂移定义为 (s9)。此处,中值取自由柱耗竭的所有hps样品。如果样品漂移不大于或小于零,则相应的中值视为0。中值柱漂移是研究中使用的所有耗竭柱的漂移中值。内源性标准化蛋白质的鉴定。下述标准用于鉴定标准化蛋白质的跃迁:(a)具有来自同一蛋白质的所有跃迁的最高中值强度;(b)在所有样品中都检测到;(c)作为标准化物在降低中值技术变异系数(cv)(即,在hps样品上测量的跃迁强度的中值cv)方面排名很高;(d)在降低样品耗竭中观察到的中值层析柱漂移方面排名很高;以及(e)具有较低的中值技术cv和较低的中值生物学cv,即在临床样品上测量的跃迁强度的中值cv。鉴定了六种内源性标准化蛋白质,并且在表33中列出。表33.内源性标准化蛋白质列表原始srm-ms数据的标准化。六个标准化跃迁用于标准化srm-ms原始数据,以降低同一研究内的样品间强度变化。对于每种样品计算比例因子,使得样品的六个标准化跃迁的强度与所有hps样品的相应中值强度保持一致。假设是样品中标准化跃迁的强度,并且是所有hps样品的相应中值强度,则关于样品的比例因子由给出,其中(s10)是强度比的中值,并且是研究中所有样品的中值。最后,对于每种样品的每个跃迁,其标准化强度计算为:(s11)其中是原始强度。通过人血浆标准(hps)样品的校准。对于无标记的ms方法,预期关于在不同实验之间的信号强度的变化。为了减少这种变化,我们利用hps样品作为外部标准,并且在发现研究和验证研究之间校准了强度。假设是样品s中对数转换(底数2)的标准化的跃迁强度,和分别是发现研究和验证研究中hps样品的相应中值。然后,hps校正强度为(s12)肽和蛋白质测定的q值计算。在srm测定的开发中,重要的是要确保检测到的跃迁对应于它们预期测量的肽和蛋白质。计算工具如mprophet(15)使得srm测定的自动化合格成为可能。我们引入了对mprophet的补充策略,其不要求对于每个数据集的定制。它利用了表达相关技术(16),以高置信度确认来自同一肽和蛋白质的跃迁的身份。在图16中,呈现了测定中的每一对跃迁之间的皮尔森相关性的直方图。一对跃迁之间的相关性得自其在发现研究中的所有样品中的表达谱。如期望的,来自相同肽的跃迁是高度相关联的。类似地,来自相同蛋白质的不同肽片段的跃迁也是高度相关联的。相比之下,来自不同蛋白质的跃迁并非高度相关联的,其使得蛋白质的srm测定质量的统计分析成为可能。为了确定假阳性测定率,我们计算了肽srm测定的q值(17)。使用来自不同蛋白质的跃迁之间的皮尔森相关分布作为零分布(图16),将经验性p值分配给在至少五个常见样品中检测到的来自同一肽的一对跃迁。如果在少于五个常见样品中检测到一对跃迁,则分配‘na’的值。使用bioconductor中的“qvalue”软件包(www.bioconductor.org/packages/release/bioc/html/qvalue.html),将经验性p值转变为q值。我们以相同的方式计算蛋白质srm测定的q值,除了将个别蛋白质的皮尔森相关性计算为来自蛋白质的不同肽的两个跃迁之间的相关性之外。对于在五个或更多个常见样品中未检测到两种肽的蛋白质,其q值无法正确地评估且分配‘na’。如果来自相同蛋白质的两种肽的跃迁的相关性高于0.5,则存在该测定为假的小于3%概率。下表中显示了36种最协同蛋白。实施例10.xl2elisa结果已开发了xpresyslung以区分良性与恶性肺部结节。xpresyslung是用于蛋白质的血液测试,其使用了大数据集组合了蛋白质组学和计算机科学方面的专业知识。质谱法已被用作分子诊断技术数十年,并且仪器中的最新进展允许一次测量数百种蛋白质。癌症分泌且脱落的蛋白质不同于正常细胞,其中这些蛋白质中的在血液中循环。从由具有良性和恶性肺部结节的两种患者贮存的388种蛋白质候选物和血液样品起始indi。最初的分析发现并验证了使用11种蛋白质的组合用于良性结节的预测物。xpresyslung版本一(xl1)提供了在临床风险因素上显著的性能,医生使用所述临床风险因素来区分良性和恶性肺部结节。indi现在已完成了关于方案收集的血液样品的进一步工作,以完善第二版xpresyslung(xl2),其为用于确定哪些结节是良性的稳固测试。这个新版本xl2在四个方面对xl1进行改善,并且这些方面是:1)改进的预期用户群体;2)鉴定先前11种蛋白质中的2种,其最准确地鉴定良性肺部结节;3)掺入五种临床危险因素;以及4)基于两项大型前瞻性研究的发现和验证,在所述研究中使用统一方案而不是档案生物银行来收集样品。xl2预期用于评估40岁或更大患者中的8-30mm肺部结节,在所述患者中,医生估计较低的癌症风险(癌症的预测概率为0至50%)。xpresyslung的目标是鉴定可能是良性的那些结节,因此这些结节可以通过ct监视安全地观察,而不是经历昂贵且有风险的侵入性程序,例如活组织检查和手术。当前的研究掺入关于xl2中使用的两种蛋白质c163a和lg3bp的结果,与elisa测量相比,使用多重反应监测质谱法(mrmms)。使用相关性和统计分析,比较来自两种技术的蛋白质测量。mrmms:通过多重反应监测质谱法(mrmms),分析在本研究中使用的十八种血浆样品。每种血浆样品被分析五次,以便生成平均xl2结果。elisa:人可溶性cd163elisa试剂盒购自通过americanresearchproductincorporated,waltham,ma02452的cuasbio,目录号csb-e14050h。人半乳凝素3bpelisa试剂盒,目录号ab213784,购自abcam,cambridge,ma02139。根据制造商的方案分析血浆样品。一式两份生成了七点标准曲线,对于人可溶性cd163蛋白,范围为100ng/ml至1.56ng/ml,且对于人半乳凝素3bp蛋白,范围为4,000pg/ml至62.5pg/ml。阴性对照也一式两份地产生。将血浆样品解冻,并且使用由每个elisa试剂盒供应的样品稀释剂稀释,以产生足够的样品体积,以一式两份评价。在将稀释的样品加入板后,使人可溶性cd163elisa板在37°c下温育2小时,并且使人半乳凝素-3bpelisa板在37°c下温育90分钟。在温育之后,弃去板内容物,并且将100μl生物素化的检测抗体加入elisa板上的每个孔中,并且在37℃下温育60分钟。在温育之后,弃去板内容物,并且用200μl适当的洗涤缓冲液将板洗涤3次。在洗涤后,将100µl抗生物素蛋白检测试剂加入每个孔中,并且对于人可溶性cd163elisa板在37°c下温育1小时,且对于人半乳凝素-3bpelisa板在37°c下温育30分钟。在温育之后,弃去板内容物,并且用200µl洗涤缓冲液将板洗涤5次。在洗涤之后,将90µltmb底物加入elisa板的每个孔中,并且使板显色15至30分钟,直到通过蓝色底物指示剂的存在,检测到足够数目的样品。然后通过向每个孔中加入100µl终止溶液以猝灭反应,来终止显色反应。然后在终止反应的30分钟内,在450nm和540nm处,在moleculardevicesspectramax190uv/vis板阅读器上读取板。在整个过程自始至终,要小心避免elisa板在洗涤或试剂添加之间变干。结果xl2被定义为:其中t=0.38且是反向评分的阈值,年龄是以岁计的受试者年龄,如果受试者是以前或当前的吸烟者,则吸烟者为1(否则为0),直径是以mm计的肺部结节的大小,如果推测为肺部结节,则毛刺为1(否则为0),并且如果肺部结节位于上肺叶,则位置为1(否则为0)。在该分析中,我们仅集中于定义为的反向评分,因为x中包含的临床因素不影响结果的比较。图17显示了mrmms和elisa数据的比较。粗水平线指示0.38的xl2阈值t。粗虚线指示elisa数据的假定阈值。左下象限和右上象限中的数据点显示mrmms和elisa方法之间的一致性。使用这两个阈值来比较结果,我们观察到16/18(89%)在两种方法之间是一致性的。关于mrmms与elisa结果之间的一致的费氏精确检验结果为p=0.0077,因此显示了一致性的显著性。实施例11.xl1和xl2替代评价测试(aat)表征研究设计定义.可接受范围:参考结果+/-3个标准差。xl1w校准的:𝑊校准的=𝑊–𝑊中值_分批_𝑝𝑐+𝑊校准因子。表征:根据至少3个等分试样的分析,确定样品的xl1w校准的和xl2反向评分的平均值和标准差。xl2反向评分:。xl1:xpresyslung测试版本1。xl2:xpresyslung测试版本2。用于表征的样品选择。选择满足下述标准的18种样品的集合用于表征。选择用于表征的样品必须具有至少1ml的残留体积,以用于在表征过程中的重复测试以及在aat事件中的将来使用。所选样品的列表包括在最终报告中。xl1样品选择。其xl1w校准的在-2.83和2.93之间(图18中的历史w校准的分布的平均值的±3标准差),在2015年6月1日后收集的先前分析样品,对于选择用于表征是合格的。xl2样品选择。其xl2反向评分在-1.08和3.49之间(图19中的历史xl2反向评分分布的平均值的±3标准差),在2015年6月1日后收集的先前分析样品,对于选择用于表征是合格的。表征过程。表征在临床lims研究中执行用于跟踪目的。选择用于表征的样品登录到lims系统中的表征临床研究内。登录每个选定样品最少七个80微升的等分试样。表征研究样品的分析遵循对于xl1测定确定的sop。每种样品的至少3个等分试样在分开的耗竭柱上以分开的分批进行加工(即,同一样品的两个等分试样不在同一批中或在同一柱上进行加工)。用于每个分批的随机化样品加工次序在样品选择后由qa生成,并且包括在最终研究报告中。表征研究的每个分批可以在用于加工商业或其它临床样品的同一耗竭柱上进行加工,然而,商业和临床研究样品不能在aat表征分批内进行加工。对至少三个等分试样的xl1w校准的求平均值,并且xl1w校准的的平均值和标准差用于确定用于aat样品档案中的适合性。结果的平均值定义了每个aat样品的参考结果。可接受的范围(w校准的的最大上限和下限[分别为w校准的,ul和ww校准的,ll)定义为在参考结果的任一侧上的三个标准差。然而,由于较小的样本大小,将w校准的的最小标准差设定为σw=0.1927476。这个最小值基于其为xpresyslung分析验证研究的部分的15个重复阳性对照样品的标准差。小于此的标准差不是预期的,并且将是在表征期间取样不足的结果。对至少三个等分试样的xl2反向评分求平均值,并且xl2反向评分的平均值和标准差用于确定用于aat样品档案中的适合性。结果的平均值定义了每个aat样品的参考结果。可接受的范围(反向评分的最大上限和下限[分别为[rsul和rsll)定义为在参考结果的任一侧上的三个标准差。然而,由于较小的样本大小,将xl2反向评分的最小标准差设定为0.216887。这个最小值基于其为xpresyslung分析验证研究的部分的15个重复阳性对照样品的标准差。小于此的标准差不是预期的,并且将是在表征期间取样不足的结果。验收标准。技术主管和质量保证(technicalsupervisorandqualityassurance)将审查最终结果,以便选择用于aat档案中的样品。为了对于aat档案合格,必须满足下述一般验收标准:(1)测试的样品必须通过如批准的sop中定义的质量控制;(2)在表征测试完成后,必须保留80微升的至少2个等分试样;以及(3)至少3个等分试样对于用于下述计算中必须是可接受的。除上述一般验收标准之外,下述验收标准应用于xl1:w校准的的最大标准差必须小于σw=0.3855。关于σw的这个最大值基于其为xpresyslung分析验证研究的部分的15个重复阳性对照样品的标准差的两倍。大于此的标准差不是预期的,并且将是在表征期间取样不足的结果。除上述一般验收标准之外,下述验收标准应用于xl2:反向评分的最大标准差必须小于σw=0.4338。关于σw的这个最大值基于其为xpresyslung分析验证研究的部分的15个重复阳性对照样品的标准差的两倍。大于此的标准差不是预期的,并且将是在表征期间取样不足的结果。样品贮存计划。选择用于aat样品档案的所有样品都贮存于-80°c冷冻库中的分开样品贮存盒中。对这个贮存的接近仅限于实验室人员和质量保证。参考文献1.albert&russellamfamphysician80:827-831(2009)2.gould等人chest132:108s-130s(2007)3.kitteringham等人jchromatrogbanalyttechnolbiomedlifesci877:1229-1239(2009)4.lange等人molsystbiol4:222(2008)5.lehtio&depetrisjproteomics73:1851-1863(2010)6.macmahon等人radiology237:395-400(2005)7.makawitaclinchem56:212-222(2010)8.ocak等人procamthoracsoc6:159-170(2009)9.ost,d.e.和m.k.gould,decisionmakinginpatientswithpulmonarynodules.amjrespircritcaremed,2012.185(4):第363-72页。10.cima,i.等人,cancergenetics-guideddiscoveryofserumbiomarkersignaturesfordiagnosisandprognosisofprostatecancer.procnatlacadsciusa,2011.108(8):第3342-7页。11.desiere,f.等人,thepeptideatlasproject.nucleicacidsres,2006.34(databaseissue):第d655-8页。12.farrah,t.等人,ahigh-confidencehumanplasmaproteomereferencesetwithestimatedconcentrationsinpeptideatlas.molcellproteomics,2011.10(9):第m110006353页。13.omenn,g.s.等人,overviewofthehupoplasmaproteomeproject:resultsfromthepilotphasewith35collaboratinglaboratoriesandmultipleanalyticalgroups,generatingacoredatasetof3020proteinsandapublicly-availabledatabase.proteomics,2005.5(13):第3226-45页。14.kearney,p.等人,proteinidentificationandpeptideexpressionresolver:harmonizingproteinidentificationwithproteinexpressiondata.jproteomeres,2008.7(1):第234-44页。15.huttenhain,r.等人,reproduciblequantificationofcancer-associatedproteinsinbodyfluidsusingtargetedproteomics.scitranslmed,2012.4(142):第142ra94页。16.henschke,c.i.等人,ctscreeningforlungcancer:suspiciousnessofnodulesaccordingtosizeonbaselinescans.radiology,2004.231(1):第164-8页。17.henschke,c.i.等人,earlylungcanceractionproject:overalldesignandfindingsfrombaselinescreening.lancet,1999.354(9173):第99-105页。18.states,d.j.等人,challengesinderivinghigh-confidenceproteinidentificationsfromdatagatheredbyahupoplasmaproteomecollaborativestudy.natbiotechnol,2006.24(3):第333-8页。19.polanski,m.和n.l.anderson,alistofcandidatecancerbiomarkersfortargetedproteomics.biomarkinsights,2007.1:第1-48页。20.krogh,a.等人,predictingtransmembraneproteintopologywithahiddenmarkovmodel:applicationtocompletegenomes.jmolbiol,2001.305(3):第567-80页。21.bendtsen,j.d.等人,improvedpredictionofsignalpeptides:signalp3.0.jmolbiol,2004.340(4):第783-95页。22.bendtsen,j.d.等人,feature-basedpredictionofnon-classicalandleaderlessproteinsecretion.proteinengdessel,2004.17(4):第349-56页。23.lange,v.等人,selectedreactionmonitoringforquantitativeproteomics:atutorial.molsystbiol,2008.4:第222页。24.picotti,p.等人,high-throughputgenerationofselectedreaction-monitoringassaysforproteinsandproteomes.natmethods,2010.7(1):第43-6页。25.mallick,p.等人,computationalpredictionofproteotypicpeptidesforquantitativeproteomics.natbiotechnol,2007.25(1):第125-31页。26.perkins,d.n.等人,probability-basedproteinidentificationbysearchingsequencedatabasesusingmassspectrometrydata.electrophoresis,1999.20(18):第3551-67页。27.hastie,t.,r.tibshirani和j.h.friedman,theelementsofstatisticallearning:datamining,inference,andprediction:with200full-colorillustrations.springerseriesinstatistics.2001,newyork:springer.xvi,第533页。28.mcclish,d.k.,analyzingaportionoftheroccurve.meddecismaking,1989.9(3):第190-5页。29.x.-j.li,c.hayward,p.-y.fong,m.dominguez,s.w.hunsucker,l.w.lee,m.mclean,s.law,h.butler,m.schirm,o.gingras,j.lamontagne,r.allard,d.chelsky,n.d.price,s.lam,p.p.massion,h.pass,w.n.rom,a.vachani,k.c.fang,l.hood和p.kearney,"ablood-basedproteomicclassifierforthemolecularcharacterizationofpulmonarynodules,"sciencetranslationalmedicine,第5卷,第207期,第207ra142页,2013。30.a.vachani,h.i.pass,w.n.rom,d.e.medthun,e.s.edell,m.laviolette,x.-j.li,p.-y.fong,s.w.hunsucker,c.hayward,p.j.mazzone,d.k.madtes,y.e.miller,m.g.walker,j.shi,p.kearney,k.c.fang和p.p.massion,"validationofamultiproteinplasmaclassifiertoidentifybenignlungnodules,"journalofthoraciconcology,第10卷,第4期,第629-637页,2015。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1