一种银杏内酯A或银杏内酯B的含量测定方法与流程
本发明属于快速分析检测领域,具体涉及近红外技术的快速分析领域,特别是涉及一种银杏叶提取、浓缩过程银杏内酯a或银杏内酯b的快速测定方法。
背景技术:
近红外(near-infrared,nir)光谱分析技术是常用的过程分析技术之一,可实现中药生产工艺的离线、在线监控与管理,其快速、无损的检测,增加了对工艺过程的了解和控制,可用于评价原料质量均一性,为过程控制提供了技术支持及数据积累平台。模型建立过程中,评价指标有相关系数(correlationcoefficient,r)、校正集误差均方根(rootmeansquareerrorofcalibration,rmsec)、交叉验证集误差均方根(rootmeansquareerrorofcrossvalidation,rmsecv)、预测集误差均方根(rootmeansquareofprediction,rmsep)、偏差(bias)、性能偏差比(ratioofperformancetodeviation,rpd)和相对标准偏差(relativestandarderror,rsd)。r越大,表明预测值与真实值越接近;rmsec、rmsecv、rmsep、bias、rsd值越小,表明模型预测准确性越高;rpd值越大,表明模型的预测能力越强。一般以rpd值≥3、rsd≤10%表示nir模型建立成功。
银杏二萜内酯葡胺注射液的主要原料为银杏内酯,是从银杏叶中提取精制得到的原料。其有效成分主要为银杏二萜内酯类成分。《中国药典》2015版规定银杏叶中萜类内酯的总量不低于0.25%,一般市场供应的银杏叶饮片中总内酯含量均值在0.3%左右,前期实验表明,银杏叶经水或乙醇煎煮后,溶液中单个萜类内酯的含量常常低于千分之一,因而也低于近红外仪器的检测限。
而且,银杏叶饮片提取或浓缩液中间体中萜类内酯的含量检测过程,样品预处理繁琐,包括加2%盐酸溶液2滴,用乙酸乙酯振摇提取4次,合并提取液,用5%醋酸钠溶液洗涤,分取醋酸钠溶液,再用乙酸乙酯10ml洗涤,合并乙酸乙酯提取液及洗涤液,用水洗涤2次,每次20ml分取水液,用乙酸乙酯10ml洗涤,合并乙酸乙酯液,回收溶剂至干,残液用甲醇溶解并转移至5ml量瓶中,加甲酵至刻度,摇匀,滤过,取续滤液等。通过上述繁琐的样品预处理过程,会增加检测误差出现概率和检测时间,不利于实现银杏叶提取、浓缩过程的快速检测及过程监控。
智能制造的大环境下,寻找实现银杏内酯a、银杏内酯b的快速检测方法对于实现银杏二萜内酯制剂产品生产过程的监控是很有必要的。
技术实现要素:
有鉴于此,本发明所要解决的技术问题是克服近红外检测限低于千分之一的缺陷,实现银杏内酯a、银杏内酯b的快速检测。
具体地,本发明提出了一种银杏叶饮片提取、浓缩过程银杏内酯a或银杏内酯b的含量测定方法,其特征在于,该方法包括,
(1)收集银杏叶提取或浓缩过程的样本;
(2)通过nir检测所述样本的固含量;
(3)根据所述固含量和银杏内酯a或b含量的关系,计算所述银杏内酯a或银杏内酯b的含量。
优选地,所述步骤(1)中,提取溶剂选自水、乙醇或乙醇水溶液。
更优选地,所述步骤(1)中,提取溶剂是水,提取温度保持在90℃以上。
具体地,所述nir检测条件为:光谱扫描起始波长1100nm,终止波长2300nm,波长增量2nm,扫描次数300次,采集模式ratio。
进一步地,所述固含量和银杏内酯a或b的量的关系通过统计学方法建立而来。比如:指标成分含量数据之间的相关性分析可以在oringin8.0软件上完成。
具体地,所述步骤(3)中,根据所述固含量和银杏内酯a或b含量的关系,计算所述银杏内酯a或银杏内酯b的含量;其中,所述固含量和银杏内酯a或b含量的关系式呈二次三项式、指数关系、对数关系。
进一步地,所述银杏内酯a或b的含量信息通过hplc法或lc-ms法获取。
本发明还提出了一种银杏内酯a或b含量与固含量关系nir测试模型的检测方法,其特征在于,该方法包括:
(1)收集银杏叶提取、浓缩过程的样本;
(2)检测所述样本的固含量,利用hplc法或lc-ms法获取样本中银杏内酯a或b的含量;
(3)拟合固含量和银杏内酯a或b含量的关系,得到nir测试模型用以对银杏内酯a或银杏内酯b含量进行检测。
具体地,所述步骤(2)中,hplc法或lc-ms法所检测的样本预处理过程包括:加2%盐酸溶液2滴,用乙酸乙酯振摇提取4次,合并提取液,用5%醋酸钠溶液洗涤,分取醋酸钠溶液,再用乙酸乙酯10ml洗涤,合并乙酸乙酯提取液及洗涤液,用水洗涤2次,每次20ml分取水液,用乙酸乙酯10ml洗涤,合并乙酸乙酯液,回收溶剂至干,残液用甲醇溶解并转移至5ml量瓶中,加甲酵至刻度,摇匀,滤过,取续滤液,即得。
进一步地,所述步骤(1)中,以重量计,提取溶剂为所述银杏叶的6-15倍量;提取溶剂是选自水或、乙醇或乙醇水溶液;提取或浓缩过程中每隔几分钟收集一定量的样本。
具体地,在固含量和银杏内酯a或b的含量的检测前,对所述步骤(1)所收集的样本进行过滤。
具体地,所述步骤(2)中,所述样本的固含量通过nir或烘干法测定。优选为通过nir测定。
进一步地,前述所用的银杏叶为经过加工或经过炮制后达到一定质量标准的饮片。
具体地,前述检测方法均用于对银杏叶提取、浓缩过程的银杏内酯a或银杏内酯b的含量进行检测。
本发明对可能用于近红外监控的指标进行了探索,并通过实验证实了固含量作为一个含量高于千分之一的指标,利用近红外技术对该指标的监控对其可实现快速、准确的监控,并基于此通过固含量的数据间接计算含量底于千分之一的银杏内酯a、银杏内酯b的含量,从而克服了近红外技术检测限的瓶颈问题,同时也降低了银杏内酯a、银杏内酯b的含量测定成本,解决了包括检测时间、样品预处理及损耗等问题,实现了银杏叶提取浓缩过程中固含量、银杏内酯a、银杏内酯b的快速监控。
附图说明
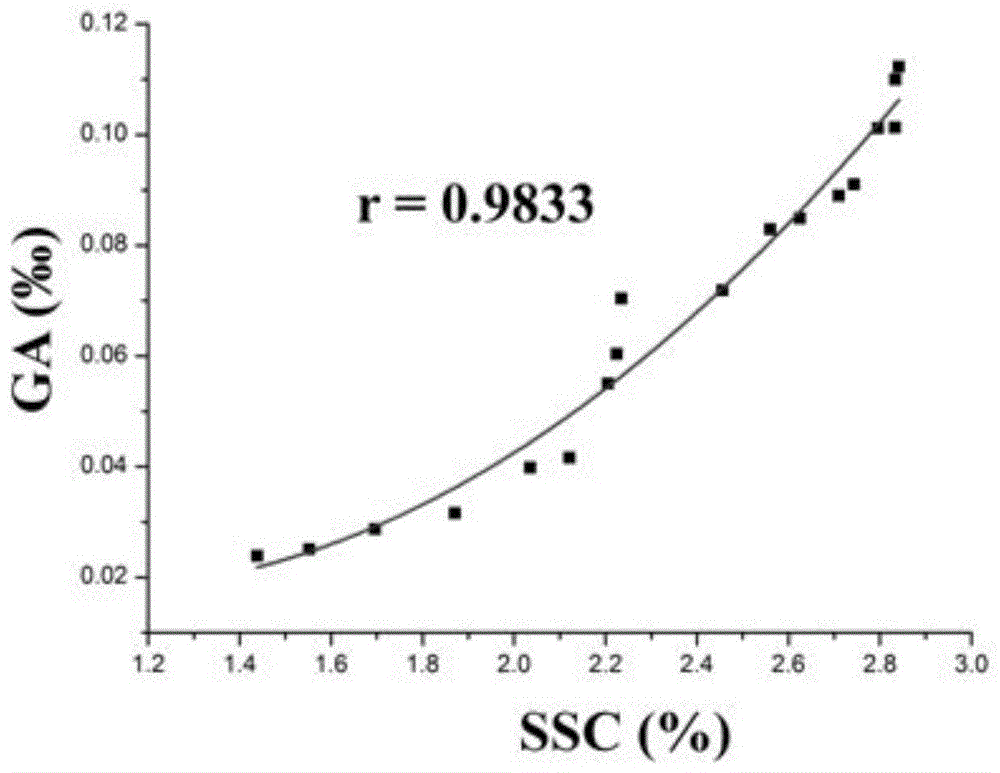
图1为银杏内酯a(ga)和固含量(ssc)关系图。
图2为银杏内酯b(gb)和固含量(ssc)关系图。
具体实施方式
本发明公开了一种银杏内酯a或银杏内酯b的快速测定方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。
以下结合具体实施例对本发明做进一步详细说明。应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制。实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
实施例1固含量、银杏内酯a或银杏内酯b近红外检测模型的建立1、样品收集
收集12批银杏叶饮片(y170101,y170202,y170301,y170309,y170401,y170501,y170601,y170702,y170801,y171102,y171201,y170104,均来自安徽广印堂中药股份有限公司),每批约取700g,置于10l实验型多功能提取罐中,加入10倍量纯化水提取,回流提取时间为2h。开始沸腾时计时,每间隔一定时间取一个样品,12批药材共收集190个样本。
2、光谱扫描
收集到的样本经过滤后要立即进行光谱的扫描,尽量模拟生产条件下光谱采集时样品的温度。打开luminar5030近红外主机,预热半小时后,点击acquire光谱采集软件,在acquisitionparameters里设置光谱采集参数如下:光谱扫描起始波长1100nm,终止波长2300nm,波长增量2nm,扫描次数300次,采集模式ratio。每个样品平行测定3次,3条光谱的平均光谱用于模型建立。
3、固含量测定
将蒸发皿置于鼓风干燥箱中进行恒重,称重m0;取5ml经离心后的一煎提取液上清液,置于已恒重的蒸发皿中,称重m1,置于水浴锅上蒸干后,再置于鼓风干燥箱中105℃进行恒重,称重m2。ssc含量的计算公式为:
4、银杏内酯a(ga)、银杏内酯b(gb)含量测定
4.1供试品溶液的制备
取提取液约50g,加2%盐酸0.5ml,用乙酸乙酯振摇提取4次(75ml、50ml、50ml、50ml),合并提取液,用5%醋酸钠溶液100ml洗涤,分取醋酸钠液,再用乙酸乙酯50ml洗涤,合并乙酸乙酯提取液及洗涤液,用水洗涤2次,每次100ml,分取水液,用乙酸乙酯50ml洗涤,合并乙酸乙酯液,回收溶剂至干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
4.2对照品溶液的制备
精密称取ga对照品10.024mg、gb对照品9.378mg,置于20ml容量瓶中,加50%丙酮溶解并定容至刻度,摇匀,即得ga浓度为0.4781mg·ml-1,gb浓度为0.4473mg·ml-1的混合对照品溶液,于4℃冰箱中冷藏,备用。
4.3色谱条件
色谱柱:kromasilc18(250×4.6mm,5μm);流动相:甲醇(25%)-四氢呋喃(10%)-水(65%);洗脱模式:等度洗脱;柱温:35℃;流速:1ml·min-1;蒸发光检测器氮气流速为2.5l·min-1;漂移管温度:105℃;分别吸取对照品溶液和供试品溶液各适量,注入液相色谱仪。
5、模型优化
将经异常点剔除、光谱预处理和变量筛选之后的样本进行回归模型的建立。采用kennard-stone法优选80%样本作为校正集样本,20%的样本作为预测集样本,采用pls法建立光谱矩阵和固含量、光谱矩阵和ga、光谱矩阵和gb之间的定量模型。
6、模型建立
表1光谱变量筛选前后建模结果
ga含量的pls模型中,其校正集rcal值为0.7650,rmsec、rmsecv和biascal值分别为0.0108、0.0124、0.0084mg·g-1;预测集rpre值为0.6083,rmsep和biaspre值分别为0.0102、0.0078mg·g-1;模型的rpd值为1.22,rsd值为17.82%。
gb含量的pls模型中,其校正集rcal值为0.7991,rmsec、rmsecv和biascal值分别为0.0062、0.0072、0.0045mg·g-1;预测集rpre值为0.7197,rmsep和biaspre值分别为0.0064、0.0053mg·g-1;模型的rpd值为1.44,rsd值为20.05%。
ssc含量的pls模型中,其校正集rcal值为0.9932,rmsec、rmsecv和biascal值分别为0.0548%、0.0718%、0.0413%;预测集rpre值为0.9962,rmsep和biaspre值分别为0.0589%、0.0460%;模型的rpd值为10.34,rsd值为2.52%。
从结果可以看出,ssc含量的pls模型的rpd>3,rsd<5%,模型建立成功,ssc可作为在线nir的监控指标。ga与gb含量的pls模型的rsd大于10%,不宜作为在线nir的监控指标和含量检测。
实施例2ga与gb含量的间接计算方法
从实施例1中可知,ga与gb含量的nir模型是检测误差大于10%,不宜用于含量的在线检测,因而进一步从指标性成分之间的相关性研究角度出发,采用lc-ms法研究不同提取时间点ga、gb含量分别与ssc含量的相关性,实现ga、gb含量的间接检测。指标成分含量数据之间的相关性分析在oringin8.0软件上完成。
1标准曲线的制备
标准品工作液:精密称取ga10.772mg,gb9.062mg,置于10ml容量瓶中,加入甲醇溶解并定容至刻度,制得每1ml含ga1.0276mg、gb0.8645mg的混合对照品储备液。取适量对照品储备液,50%甲醇逐倍稀释,制得系列混合工作液。
内标溶液的制备:精密称取木犀草素10.024mg,置于10ml容量瓶中,加入甲醇溶解,定容,即得浓度为0.9983mg·ml-1的储备液,临用前以50%甲醇稀释成500ng·ml-1的内标工作液。
2样本预处理
精密称量约0.4g的不同时间点的提取液(样本来自于实施例1),置于10ml容量瓶中,50%甲醇定容,微孔滤膜过滤(0.22μm),移液枪精密量取续滤液100μl,加50%甲醇稀释至600μl,涡旋,精密量取200μl稀释液并加入50μl木犀草素(内标),涡旋即得供试品溶液。
3含量测定
通过实施例1第3部分固含量检测方法获取样品的固含量数据,样品中ga、gb的含量测定条件如下所示:
色谱条件:
色谱柱:agilentporoshellsb-c18(4.6×150mm,2.7μm);流动相:乙腈(42)-0.1%甲酸(58);等度洗脱;时间3.5min;柱温:35℃;进样量5ul;流速:0.3ml·min-1。
质谱条件:
质谱扫描方式为多重离子反应监测(mrm)方式,检测条件见表2。
表2ga和gb的质谱检测参数
以对照品浓度(ng·ml-1)为横坐标(x),峰面积为纵坐标(y),绘制回归方程。ga的方程式为y=0.0001x-0.0013(r=0.9998),gb的方程式为y=0.005x+0.1718(r=0.9974),结果表明ga浓度在21.544-2154.4ng·ml-1、gb浓度在18.124-1812.4ng·ml-1之间呈现良好的线性关系。
4相关性拟合结果
对样品含量数据进行相关性分析,二次多项式拟合结果如图1和图2所示。ga-ssc含量间的关系式为y=0.0489-0.05882x+0.02781x2(r=0.9833);gb-ssc含量间的关系式为y=0.00717-0.01414x+0.01162x2(r=0.9871)。不同时间点的ga-ssc与gb-ssc含量之间存在相关性,关系式呈二次三项式,根据固含量含量,来计算ga、gb的含量。
5间接计算结果验证
取银杏叶饮片(批号:y180101,安徽广印堂中药股份有限公司)约700g,置于10l实验型多功能提取罐中,加入纯化水10倍量进行提取,回流提取时间为2h。开始沸腾时计时,每间隔一定时间取一个样品,共收集新样本16个。通过实施例1固含量的nir模型获取样品的固含量数据,根据相关性拟合关系式间接计算ga、gb的含量,ga或gb含量间接计算方法的验证结果如表3所示。
表3ga或gb含量间接计算方法的验证
经含量测定测得ga或gb的真实值,和利用近红外测得固含量后计算所得ga或gb的预测值相比,rsd均值在5%以内,预测值与真实值之间无显著差异,表明本发明建立的间接计算银杏内酯a或银杏内酯b含量的方法准确、可靠。
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!