一种3,4-亚甲二氧基乙卡西酮的检测方法与流程
本发明属于毒品司法检测领域,具体涉及一种3,4-亚甲二氧基乙卡西酮的检测方法。
背景技术:
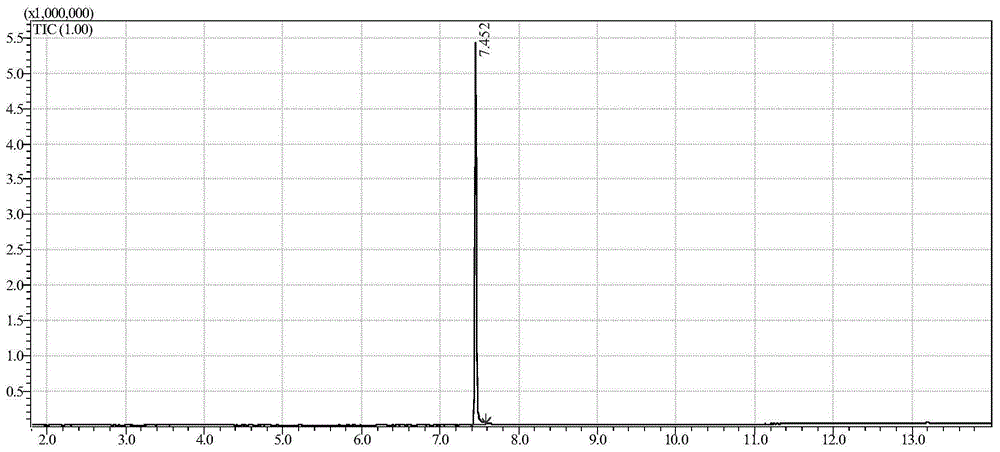
:新精神活性物质,英文名称newpsychoactivesubstances,简称nps,早期叫作legalhighs或者designerdrug,中文称“合法兴奋剂”或“策划药物”,是研究生产者为了规避打击而对部分管制毒品进行化学结构修饰得到的毒品类似物,具有与管制毒品相似的作用,有的兴奋、致幻、麻醉效果更强。联合国毒品与犯罪办公室(unodc)将新精神活性物质分为9个大类,分别是:合成大麻素类、卡西酮类、苯乙胺类、色胺类、哌嗪类、氯胺酮及苯环利定类、氨基茚类、植物类和其他类。其中合成大麻素类物质和卡西酮类物质是滥用最为严重的新精神活性物质。3,4-亚甲二氧基乙卡西酮属于卡西酮类物质,于2015年10月被列入《非药用类麻醉药品和精神药品管制品种增补目录》进行列管,属于禁售药品。目前我国对于新精神活性物质的研究还很匮乏,针对已管制的3,4-亚甲二氧基乙卡西酮定性定量的检测方法还是空白,因此亟须建立其定性定量检测方法,为相关案件的侦查起诉做好技术支持。技术实现要素:本发明的一个目的是提供一种对疑似毒品样品中的3,4-亚甲二氧基乙卡西酮(以下简称ethylone)的气相色谱-质谱联用(gc-ms)的检测方法。本发明所提供的对疑似毒品样品中的3,4-亚甲二氧基乙卡西酮(以下简称ethylone)的气相色谱-质谱联用(gc-ms)的检测方法,包括下述步骤:1)对疑似毒品样品中的3,4-亚甲二氧基乙卡西酮进行有机溶剂提取,收集提取液;2)对所述提取液利用气相色谱-质谱联用进行定性检测;将所述提取液与3,4-亚甲二氧基乙卡西酮标准物质的总离子流色谱图和质谱图进行比对,同时满足如下两个条件时,判定为检出3,4-亚甲二氧基乙卡西酮;否则判定为未检出:a)所述提取液中目标物的保留时间与3,4-亚甲二氧基乙卡西酮标准物质的保留时间的相对误差小于2%;b)所述提取液中目标物扣除背景后的质谱图与3,4-亚甲二氧基乙卡西酮标准物质扣除背景后的质谱图匹配度大于90%。上述的方法中,当步骤2)中判定为检出3,4-亚甲二氧基乙卡西酮时,所述方法还包括对所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮进行定量分析的步骤;所述定量分析包括如下步骤:(3)将所述提取液用气相色谱进行检测;(4)采用外标法对所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮的含量进行定量分析。上述方法步骤1)中,所述有机溶剂具体可为甲醇,所述提取在超声条件下进行。上述方法步骤2)中,所述气相色谱-质谱联用(gc-ms)的检测条件可参考下述条件,当然也可根据不同品牌仪器和不同样品等实际情况进行调整。a)仪器:gc-ms。b)离子源:电子轰击源(ei)。c)质量范围:35amu~500amu。d)采集方式:全扫描(scan)。e)色谱柱类型:db-5ms石英玻璃毛细柱(5%苯基+95%聚二甲基硅氧烷)或其他等效柱。f)色谱柱参数:30m×0.25mm×0.25μm。g)色谱柱温程:140℃(3min)-20℃/min-300℃(16min)。h)进样口温度:280℃。i)传输线温度:250℃。j)离子源温度:230℃。k)分流比:40:1。l)载气:高纯氦气(he)。m)柱流量(恒流):1.0ml/min。n)倍增器电压:参考调谐状况。o)溶剂切割:2min。上述方法步骤3)中,所述气相色谱的检测条件参考如下条件,具体可根据不同品牌仪器和不同样品等实际情况进行调整。a)仪器:气相色谱仪;b)检测器:火焰离子化检测器(fid);c)色谱柱型号:db-5石英玻璃毛细柱(5%苯基+95%聚二甲基硅氧烷)或其他等效柱;d)色谱柱参数:30m×0.25mm×0.25μm。e)柱温:140℃(3min)-20℃/min-300℃(16min)。f)进样口温度:280℃。g)检测器温度:300℃。h)载气:高纯氮气(n2)。i)分流比:20:1。j)柱流速(恒流):1ml/min。k)燃烧气:h2。l)燃烧气流速:按仪器默认值。m)助燃气:空气。n)助燃气流速:按仪器默认值。上述方法步骤4)中,所述定量分析具体包括如下步骤:1)配制至少5个不同浓度的3,4-亚甲二氧基乙卡西酮标准溶液,将所述标准溶液采用步骤3)中所述的气相色谱进行检测,得到不同浓度的3,4-亚甲二氧基乙卡西酮的色谱峰面积,以所述标准溶液的浓度为横坐标,以峰面积为纵坐标,制作标准工作曲线并得到回归方程;2)将步骤3)测定得到的所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮的峰面积带入所述回归方程中,计算得到所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮的含量;所述3,4-亚甲二氧基乙卡西酮标准溶液的浓度范围为0.05~2.5mg/ml。本发明的另一个目的是提供一种对疑似毒品样品中的3,4-亚甲二氧基乙卡西酮(以下简称ethylone)用液相色谱-串级质谱联用(lc-ms/ms)在多反应监测(mrm)模式下进行检测的方法。本发明所提供的对疑似毒品样品中的3,4-亚甲二氧基乙卡西酮(以下简称ethylone)用液相色谱-串级质谱联用(lc-ms/ms)在多反应监测(mrm)模式下进行检测的方法,包括下述步骤:a)对疑似毒品样品中的3,4-亚甲二氧基乙卡西酮进行有机溶剂提取,收集提取液;b)采用液相色谱-串级质谱联用(lc-ms/ms)在多反应监测(mrm)模式下对所述提取液进行定性检测;在相同的试验条件下,所述提取液中出现两对定性离子对色谱峰,其保留时间与3,4-亚甲二氧基乙卡西酮标准物质保留时间比较,相对误差在±2%内,且相对离子对丰度比与3,4-亚甲二氧基乙卡西酮标准物质中的相对离子对丰度比之相对误差不超过最大允许相对误差规定的范围,则判断样品中存在3,4-亚甲二氧基乙卡西酮;所述3,4-亚甲二氧基乙卡西酮的两对定性离子对为:1)m/z222.1/174.1,去簇电压为30v,碰撞能量为17ev;2)m/z222.1/146.1,去簇电压为30v,碰撞能量为25ev;相对离子对丰度比的最大允许相对误差的规定如下:上述的方法中,当步骤b)中判定为检出3,4-亚甲二氧基乙卡西酮时,所述方法还包括对所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮进行定量分析的步骤;所述定量分析包括如下步骤:c)将所述提取液用液相色谱进行检测;d)采用外标法对所述疑似毒品样品中3,4-亚甲二氧基乙卡西酮的含量进行定量分析。上述方法步骤a)中,所述有机溶剂具体可为甲醇,所述提取在超声条件下进行。上述方法步骤b)中,所述液相色谱-串级质谱的色谱条件如下,可根据不同品牌仪器和不同样品等实际情况进行调整:a)进样体积:1μl。b)色谱柱:c18液相色谱柱(1.7μm,100mm×2.1mm)或其他等效柱;c)流动相:a体积分数0.1%甲酸水溶液,b乙腈。d)梯度程序:0~1.5min,a相体积分数为98%、b相体积分数为2%;1.5~6.5min,a相体积分数从98%降至10%、b相体积分数从2%升至90%;6.5~8.0min,a相体积分数为10%、b相体积分数为90%;8.0~8.1min,a相体积分数从10%升至98%,b相体积分数从90%降至2%;8.1~10.0min,a相体积分数为98%、b相体积分数为2%;e)流速:0.4ml/min。f)离子源:电喷雾离子源-正离子模式(esi+)。g)检测方式:多反应监测(mrm)。h)离子源温度:550℃。i)离子喷雾电压:5500v。j)碰撞气(cad)、气帘气(cur)、雾化气(gs1)、辅助气(gs2)均为高纯氮气,使用前调节各气流流量以使质谱灵敏度达到检测要求。k)去簇电压(dp)、碰撞能量(ce)应优化至最佳灵敏度。上述方法步骤c)中,所述液相色谱的色谱条件如下,可根据不同品牌仪器和不同样品等实际情况进行调整:a)色谱柱:c18色谱柱(5μm,150mm×4.6mm)或其他等效柱。b)柱温:35℃。c)流速:1.0ml/min。d)进样体积:5.0μl。e)检测波长:280nm,带宽=4nm,参比波长=400nm,参比带宽=100nm。f)流动相:a相为乙腈,b相为磷酸-三乙胺缓冲液(ph2.6)。g)洗脱程序:选择等度洗脱或梯度洗脱;所述样品为纯度较高(浓度范围是70%~100%),则采用等度洗脱,流动相由a相和b相按照体积比14:86的比例组成,分析时间8min;所述样品为成分复杂的样品,则采用梯度洗脱,梯度程序为:0~7.5min,a相体积分数为14%、b相体积分数为86%;7.5~7.7min,a相体积分数从14%升至80%、b相体积分数从86%降至20%;7.7~9.5min,a相体积分数为80%、b相体积分数为20%;9.5~9.7min,a相体积分数从80%降至14%,b相体积分数从20%升至86%;9.7~15min,a相体积分数为14%、b相体积分数为86%;h)检测器:dvd或uv检测器;上述的方法步骤d)中,所述定量分析具体包括如下步骤:1)配制至少5个不同浓度的3,4-亚甲二氧基乙卡西酮标准溶液,将所述标准溶液采用步骤c)中所述的液相色谱条件进行检测,得到不同浓度的3,4-亚甲二氧基乙卡西酮的色谱峰面积,以所述标准溶液的浓度为横坐标,以峰面积为纵坐标,制作标准工作曲线并得到回归方程;2)将步骤c)测定得到的所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮的峰面积带入所述回归方程中,计算得到所述疑似毒品样品中的3,4-亚甲二氧基乙卡西酮的含量;所述3,4-亚甲二氧基乙卡西酮标准溶液的浓度范围为0.005~0.5mg/ml。上述方法中,对样品溶液(提取液)进行稀释的方法,是将样品溶液与磷酸-三乙胺缓冲液按1:4(v/v)比例进行稀释。与现有技术相比,本发明具有如下优点:利用常用的气相色谱-质谱联用、气相色谱、液相色谱-串级质谱、液相色谱技术建立3,4-亚甲二氧基乙卡西酮的定性定量分析方法,所建立的方法快捷、简便、易于操作,便于推广应用,能够满足司法鉴定部门对此类案件办理的需要,为该类案件的顺利诉讼提供依据。附图说明图1为气相色谱-质谱分析ethylone(rt=7.45min)得到的总离子流色谱图。图2为ethylone参考ei质谱图(电子轰击电离质谱)。图3为气相色谱分析ethylone(rt=8.18min)得到的色谱图。图4为液相色谱-串联质谱分析ethylone得到的mrm色谱图(m/z222.1/174.1;m/z222.1/146.1)。图5为液相色谱分析ethylone(rt=3.73min)得到的色谱图。具体实施方式下面通过具体实施例对本发明的方法进行说明,但本发明并不局限于此,凡在本发明的精神和原则之内所做的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。下述实施例中的定量试验,均设置三次重复实验,结果取平均值。实施例1、对疑似毒品样品中的ethylone用气相色谱-质谱联用(gc-ms)进行定性分析1、试剂及标准物质、仪器及量器具1.1试剂及标准物质本法所用试剂均为分析纯。提取试剂:甲醇。可溯源标准物质:ethylone。1.2仪器及量器具气相色谱仪(gc),配有火焰离子化检测器(fid)。气相色谱-质谱联用仪(gc-ms),配有标准电子轰击源(ei)的四极杆质谱仪。离心机。电子天平,实际分度值d=0.01mg。振荡器。10ml和1ml移液器或移液管。10ml瓶口移液器。注:所涉及所有仪器、量器具需经过计量检定合格。2定性分析2.1样品制备样品充分研磨混匀,称取5mg置于离心管中,加入10ml甲醇,超声溶解,涡旋混匀,0.45μm滤膜过滤,待分析用。如果样品中目标物浓度过低,可增加称样量至100mg。2.2气相色谱-质谱联用(gc-ms)参考条件以下为参考条件,可根据不同品牌仪器和不同样品等实际情况进行调整。a)仪器:gc-ms。b)离子源:电子轰击源(ei)。c)质量范围:35amu~500amu。d)采集方式:全扫描(scan)。e)色谱柱类型:db-5ms石英玻璃毛细柱(5%苯基+95%聚二甲基硅氧烷)或其他等效柱。f)色谱柱参数:30m×0.25mm×0.25μm。g)色谱柱温程:140℃(3min)-20℃/min-300℃(16min)。h)进样口温度:280℃。i)传输线温度:250℃。j)离子源温度:230℃。k)分流比:40:1。l)载气:高纯氦气(he)。m)柱流量(恒流):1.0ml/min。n)倍增器电压:参考调谐状况。o)溶剂切割:2min。3定性结果评价将样品与ethylone标准物质的总离子流色谱图(见图1)和质谱图(见图2)进行比对,同时满足如下两个条件时,判定为检出相应的物质;否则判定为未检出:a)样品中目标物的保留时间与标准物质的保留时间的相对误差小于2%;b)样品中目标物扣除背景后的质谱图与标准物质扣除背景后的质谱图匹配度大于90%。实施例2、气相色谱-氢火焰离子化检测器(gc-fid)对疑似毒品样品中的ethylone进行定量分析1、试剂及标准物质、仪器及量器具1.1试剂及标准物质本法所用试剂均为分析纯。提取试剂:甲醇。可溯源标准物质:ethylone。1.2仪器及量器具气相色谱仪(gc),配有火焰离子化检测器(fid)。气相色谱-质谱联用仪(gc-ms),配有标准电子轰击源(ei)的四极杆质谱仪。离心机。电子天平,实际分度值d=0.01mg。振荡器。10ml和1ml移液器或移液管。10ml瓶口移液器。注:所涉及所有仪器、量器具需经过计量检定合格。2定量分析2.1外标单点法2.1.1标准溶液的配制2.1.1.11.0mg/mlethylone标准储备液的配制准确称取一定量的ethylone标准物质,用甲醇溶解,配制成1.0mg/mlethylone标准储备液,置冰箱冷藏保存,保存时间为6个月。2.1.1.20.1mg/mlethylone标准工作液的配制准确移取适量1.0mg/mlethylone标准储备液,用甲醇稀释至0.1mg/ml,振荡混匀,置冰箱中冷藏保存,保存时间为3个月。2.1.2气相色谱(gc-fid)参考条件以下为参考条件,可根据不同品牌仪器和不同样品等实际情况进行调整。a)仪器:gc。b)检测器:火焰离子化检测器(fid)。c)色谱柱型号:db-5石英玻璃毛细柱(5%苯基+95%聚二甲基硅氧烷)或其他等效柱。d)色谱柱参数:30m×0.25mm×0.25μm。e)柱温:140℃(3min)-20℃/min-300℃(16min)。f)进样口温度:280℃。g)检测器温度:300℃。h)载气:高纯氮气(n2)。i)分流比:20:1。j)柱流速(恒流):1ml/min。k)燃烧气:h2。l)燃烧气流速:按仪器默认值。m)助燃气:空气。n)助燃气流速:按仪器默认值。2.1.3含量范围预分析预分析的目的在于要对样品中的ethylone含量进行初测,以计算准确定量分析时所用样品的称量重量,从而保证准确定量时样品溶液中目标物的最终浓度尽可能接近所使用标准工作液的浓度。称量约5mg样品置于带盖试管中,加入10ml甲醇,超声振荡使其溶解,离心后取上清液,用gc-fid分析,采用0.1mg/ml的ethylone标准工作液作为定量参照。如果样品溶液中目标化合物的浓度过高,可适当减少样品称量重量,或将样品提取液用提取溶剂稀释后进行分析。如果样品溶液中目标化合物的浓度过低,可适当增加样品称量重量,或将样品提取液挥干、定容后进行分析。按照下列公式1计算样品中目标组分的百分含量。式中:w(%)——样品中ethylone的百分含量;c0——标准工作液中ethylone的浓度,mg/ml;a——样品溶液中ethylone的色谱峰面积值;v1——样品溶液初次定容体积,ml;v3——将v2稀释时加入提取溶剂的体积,ml;a0——标准工作液中ethylone的色谱峰面积值;v2——从v1中移取样品溶液的体积,ml;m——用于测定的样品的称量重量,mg。2.1.4准确定量2.1.4.1称量重量的计算根据预分析中ethylone含量测定结果,选择合适的定容体积、稀释倍数(参见表1),并按照下列公式2计算出样品称量重量(m)。式中:m(mg)——称量重量;c0——标准工作液中ethylone的浓度,mg/ml;v——初次定容体积,ml;d——稀释倍数;w(%)——样品中ethylone的预分析百分含量。表1外标定量法样品提取操作参数选择表2.1.4.2样品提取根据2.1.4.1计算结果,平行称取样品2份,根据预分析含量范围,加入相应初次定容体积的甲醇(参见表1),密封并超声振荡10min;如果样品溶液需要稀释,移取1ml样品提取液至另一试管中,按照稀释倍数进行稀释定容,离心后取上清液用gc-fid分析。2.1.4.3含量计算按照2.1.3中公式1计算样品中ethylone百分含量。2.2外标工作曲线法分别取低(0.05mg/ml)、中(1.0mg/ml)、高(2.5mg/ml)3个浓度的ethylone标准溶液,各浓度连续进样10针,计算样本的日内相对标准偏差;连续检测6天,计算样本的日间相对标准偏差。ethylone的日内相对标准偏差为1.90%,日间相对标准偏差为2.32%。检出限为10μg/ml。采用空白加样回收的方法,向空白溶液中分别添加不同量的ethylone标准溶液,得到浓度分别为0.05mg/ml、1.0mg/ml、2.5mg/ml的ethylone溶液各3份,测得ethylone的准确度范围为97.18%-102.32%。2.2.1标准工作曲线绘制准确称取一定量的ethylone标准物质,用甲醇溶解,配成含量为2.5mg/ml的标准贮备液,0~5℃冰箱中密封保存,有效期6个月。准确移取适量ethylone标准贮备液,依次用甲醇进行稀释到1、0.5、0.2、0.1、0.05mg/ml浓度,充分混匀。采用2.5、1、0.5、0.2、0.1、0.05mg/ml等6个浓度绘制成线性范围为0.05~2.5mg/ml的ethylone标准工作曲线。每个浓度进样3次,并对3次峰面积的平均值a及质量浓度c(mg/ml)进行线性回归,得到ethylone的回归方程:a=590.55c-5.6514,r2为1,线性范围0.05~2.5mg/ml,检出限10μg/ml(s/n≥3)2.2.2定量2.2.2.1提取操作平行称取样品2份各约25mg,加入10ml甲醇,超声溶解,离心后用gc-fid分析,采用2.2.1中的标准工作曲线进行定量计算。2.2.2.2气相色谱(gc-fid)参考条件同2.1.2外标单点定量法gc-fid参考条件。2.2.2.3含量计算根据工作曲线计算样品含量。3定量结果评价3.1含量结果有效性对同一份样品,2个平行测定数据按照公式3进行相对相差(rd)计算,rd不超过10%,数据有效;否则,应重新检验。式中:rd-相对相差,单位为%;x1、x2-两个样品平行定量测定的含量数值;-两个样品平行定量测定含量的平均值。3.2含量结果计算以2份样品测定含量的平均值作为含量结果。3.3含量结果表述定量检验完成后,检验结果应表述为:从样品中检出ethylone成分,其中ethylone(c12h15no3)含量为××.×%。本实施例中ethylone标准物质的气相色谱图如图3所示,其中保留时间为8.18min处的吸收峰为ethylone吸收峰。实施例3、对疑似毒品样品中的ethylone用液相色谱-串级质谱联用(lc-ms/ms)在多反应监测(mrm)模式下进行定性分析1、试剂及标准物质、仪器及量器具1.1试剂及标准物质所用试剂均为色谱纯,试验用水为一级水(见gb/t6682-2008),试剂及标准物质包括:a)试剂:甲醇,乙腈,甲酸;b)可溯源标准物质:ethylone;c)0.1%甲酸水溶液:量取1ml甲酸,加入水溶解并稀释至1000ml,混匀后0.22μm滤膜过滤,超声后静置待用;d)标准储备液:称取ethylone标准物质,分别用甲醇溶解,配制成2.5mg/ml的标准储备液,置冰箱冷藏保存,保存时间为6个月;e)定性用100ng/ml标准工作液:准确移取适量ethylone标准储备液,用0.1%甲酸水溶液逐级稀释至100ng/ml,待分析用,即配即用;1.2仪器及量器具仪器及量器具包括:a)液相色谱-串级质谱联用仪(lc-ms/ms),配有电喷雾离子源的质谱仪。b)离心机。c)电子天平,实际分度值d=0.01mg。d)振荡器。e)10ml和1ml移液器或移液管。f)10ml瓶口移液器。g)0.22μm滤膜。2、定性分析2.1样品制备称取样品约5mg,加入10ml甲醇,超声溶解,涡旋混匀。取适量样品溶液,用0.1%甲酸水溶液逐级稀释至100ng/ml,0.22μm滤膜过滤,待分析用,即配即用。如果样品中目标物浓度过低,可稀释至5000ng/ml。2.2液相色谱-质谱联用仪参考条件以下为参考条件,可根据不同品牌仪器和不同样品等实际情况进行调整:a)进样体积:1μl。b)色谱柱:c18液相色谱柱(1.7μm,100mm×2.1mm)或其他等效柱。c)流动相:a0.1%甲酸水溶液,b乙腈。d)梯度程序:见表2。表2梯度洗脱程序时间/mina%b%09821.59826.510908.010908.198210982e)流速:0.4ml/min。f)离子源:电喷雾离子源-正离子模式(esi+)。g)检测方式:多反应监测(mrm)。h)离子源温度:550℃。i)离子喷雾电压:5500v。j)碰撞气(cad)、气帘气(cur)、雾化气(gs1)、辅助气(gs2)均为高纯氮气,使用前调节各气流流量以使质谱灵敏度达到检测要求。k)去簇电压(dp)、碰撞能量(ce)应优化至最佳灵敏度。l)ethylone的定性离子对、去簇电压和碰撞能量对见表3。表3ethylone的定性离子对、去簇电压(dp)和碰撞能量(ce)3定性结果评价在相同的试验条件下,待测样品中出现两对定性离子对色谱峰,其保留时间与添加样品中目标物保留时间比较,相对误差在±2%内,且相对离子对丰度比与添加样品中的相对离子对丰度比之相对误差不超过表4规定的范围,则可判断样品中存在目标物。表4相对离子对丰度比的最大允许相对误差(%)实施例4、液相色谱-二级管阵列或紫外检测器(lc-dvd或lc-uv)对疑似毒品样品中的ethylone进行定量分析1、试剂及标准物质、仪器及量器具1.1试剂及标准物质所用试剂均为色谱纯,试验用水为一级水(见gb/t6682-2008),试剂及标准物质包括:a)试剂:甲醇,乙腈,甲酸,磷酸,三乙胺;b)可溯源标准物质:ethylone;c)磷酸-三乙胺缓冲液:量取浓磷酸4.12ml,加入200ml水稀释;量取三乙胺5.56ml,滴入上述磷酸溶液中;再加入水稀释至1000ml,混匀后0.45μm水系滤膜过滤,超声后静置待用;d)甲醇-磷酸-三乙胺缓冲液混合溶液(v/v=1:4):量取10ml甲醇和40ml磷酸-三乙胺缓冲液,混匀,超声后静置待用;e)标准储备液:称取ethylone标准物质,分别用甲醇溶解,配制成2.5mg/ml的标准储备液,置冰箱冷藏保存,保存时间为6个月;f)外标定量用0.1mg/ml标准工作液:移取2.5mg/ml标准储备液2ml加入到50ml容量瓶中,用甲醇稀释至刻度,配制成0.1mg/ml目标物标准工作液,置冰箱中冷藏保存,保存时间为3个月。1.2仪器及量器具仪器及量器具包括:a)液相色谱仪(lc),配有二极阵列或紫外检测器的液相色谱仪。b)离心机。c)电子天平,实际分度值d=0.01mg。d)振荡器。e)10ml和1ml移液器或移液管。f)10ml瓶口移液器。g)0.45μm滤膜。2定量分析2.1液相色谱仪参考条件以下为参考条件,可根据不同品牌仪器和不同样品等实际情况进行调整:a)色谱柱:c18色谱柱(5μm,150mm×4.6mm)或其他等效柱。b)柱温:35℃。c)流速:1.0ml/min。d)进样体积:5.0μl。e)检测波长:280nm,带宽=4nm,参比波长=400nm,参比带宽=100nm。f)流动相:a相为乙腈,b相为磷酸-三乙胺缓冲液(ph2.6)。g)洗脱程序:根据样品情况选择等度洗脱或梯度洗脱。等度洗脱(适用于纯度较高的样品):14%a+86%b(v/v),分析时间8min。梯度洗脱(适用于成分较为复杂的样品):洗脱程序参考表5。表5梯度洗脱程序时间/mina%b%014867.514867.780209.580209.714861514862.2外标单点法2.2.1含量范围预分析预分析的目的在于要对样品中的目标物含量进行初测,以计算准确定量分析时所用样品的称量重量,从而保证准确定量时样品溶液中目标物的最终浓度尽可能接近所使用标准工作液的浓度。称量约5mg样品置于带盖试管中,加入10ml甲醇振荡使其溶解,离心后取适量上清液,按样品溶液:磷酸-三乙胺缓冲液=1:4(v/v)的比例进行稀释,混匀后0.45μm有机系滤膜过滤,用lc-dvd或lc-uv分析。采用0.1mg/ml的标准工作液作为定量参照,标准溶液采用同样方式稀释,移取0.1mg/ml标准工作液适量,按标准工作液:磷酸-三乙胺缓冲液=1:4(v/v)的比例进行稀释,充分混匀,待分析用,即配即用。如果样品溶液中目标化合物的浓度过高,可适当减少样品称量重量,或将样品提取液用提取溶剂稀释后进行分析。如果样品溶液中目标化合物的浓度过低,可适当增加样品称量重量,或将样品提取液挥干、定容后进行分析。按照公式(1)计算样品中目标组分的百分含量。式中:w——样品中ethylone的百分含量,单位为%;c0——标准工作液中ethylone的浓度,单位为mg/ml;a——样品溶液中ethylone的色谱峰面积值;v1——样品溶液初次定容体积,单位为ml;v3——将v2稀释时加入提取溶剂的体积,单位为ml;a0——标准工作液中ethylone的色谱峰面积值;v2——从v1中移取样品溶液的体积,单位为ml;m——用于测定的样品的称量重量,单位为mg。2.2.2准确定量2.2.2.1称量重量的计算根据预分析中目标物含量测定结果,选择合适的定容体积、稀释倍数(样品提取操作参数选择参见表1),并按照公式(2)计算出样品称量重量(m)。式中:m——称量重量,单位为mg;c0——标准工作液中ethylone的浓度,单位为mg/ml;v——初次定容体积,单位为ml;d——稀释倍数;w——样品中ethylone的预分析百分含量,单位为%。2.2.2.2样品提取根据2.2.2.1计算结果,平行称取样品2份,根据预分析含量范围,加入相应初次定容体积的甲醇。密封并振荡10min。如果样品溶液需要稀释,移取1ml样品提取液至另一试管中,按照稀释倍数进行稀释定容,离心后取上清液,按样品溶液:磷酸-三乙胺缓冲液=1:4(v/v)的比例进行稀释,用lc-dvd(或lc-uv)分析。样品提取操作参数选择参见表1。2.2.2.3含量计算按照按照2.2.1中公式(1)计算样品中目标物百分含量。2.3外标工作曲线法分别取低(0.005mg/ml)、中(0.1mg/ml)、高(0.5mg/ml)3个浓度的ethylone标准溶液,各浓度连续进样10针,计算样本的日内相对标准偏差;连续检测6天,计算样本的日间相对标准偏差。ethylone的日内相对标准偏差为1.51%,日间相对标准偏差为2.13%。检出限为0.16μg/ml。采用空白加样回收的方法,向空白溶液中分别添加不同量的ethylone标准溶液,得到浓度分别为0.005mg/ml、0.1mg/ml、0.5mg/ml的ethylone溶液各3份,测得ethylone的准确度范围为96.55%-100.33%。2.3.1标准工作曲线绘制准确移取适量ethylone标准储备液,按标准储备液:磷酸-三乙胺缓冲液=1:4(v/v)的比例进行稀释,充分混匀,配制成0.5mg/ml的标准工作溶液;再使用甲醇-磷酸-三乙胺缓冲液混合溶液对以上0.5mg/ml的标准工作溶液进行稀释,配制成浓度为0.005mg/ml、0.025mg/ml、0.05mg/ml、0.1mg/ml、0.25mg/ml、0.5mg/ml的标准系列工作溶液。标准系列工作溶液于冰箱中冷藏保存,有效期7天。每个浓度进样3次,并对3次峰面积的平均值a及质量浓度c(mg/ml)进行线性回归,得到ethylone的回归方程:a=6099.3c+19.535,r2为0.9999,线性范围0.005~0.5mg/ml,检出限0.16μg/ml(s/n≥3)。2.3.2定量2.3.2.1提取操作平行称取样品2份各约25mg,加入10ml甲醇,超声溶解,涡旋混匀。取适量样品溶液,按样品溶液:磷酸-三乙胺缓冲液=1:4(v/v)的比例进行稀释,混匀后0.45μm有机系滤膜过滤,用lc-dvd(或lc-uv)分析。2.3.2.2含量计算根据2.3.1中的标准工作曲线进行定量,计算样品含量。3定量结果评价3.1含量结果有效性对两个平行测定数据按照公式(4)进行相对相差(rd)计算,rd不超过10%,数据有效;否则,应重新检验。式中:rd-相对相差,单位为%;x1、x2-两个样品平行定量测定的含量数值;-两个样品平行定量测定含量的平均值。3.2含量结果计算以两份样品测定含量的平均值作为含量结果。3.3含量结果表述定量检验完成后,检验结果应表述为:从样品中检出ethylone成分,其中ethylone(c12h15no3)含量为××.×%。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1