一种铁矿石中全铁含量的测定方法与流程
本发明属于铁含量分析领域,具体涉及一种铁矿石中全铁含量的测定方法。
背景技术:
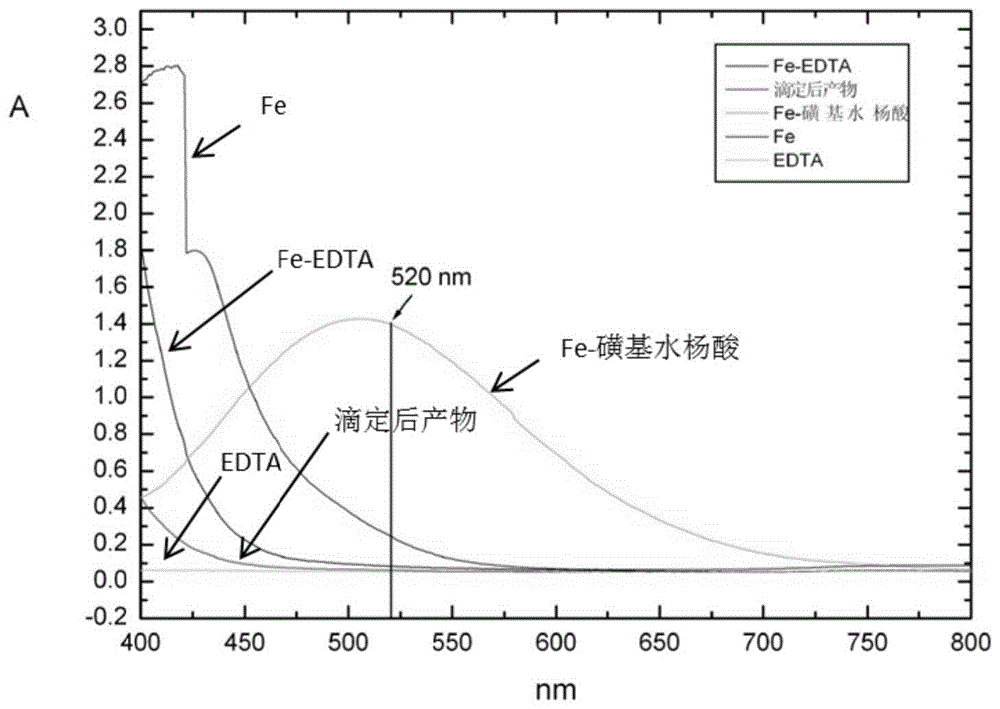
:随着中国铁矿石进口量的增加和国际铁矿石价格的飞涨,给检验检疫工作带来了更大的挑战。据统计,2018年中国铁矿石总进口量创纪录达到10.64亿吨。由于铁矿价格波动大,并且贸易双方均以总铁作为计价元素,常常出现双方总铁含量的结果不同而产生争议,提高测定方法的准确度具有重要的意义。同时,为加快货物的运转,必须采取快速而简便的检测方法对铁矿进行检验。目前,铁矿石中总铁的分析方法较多,主要有氯化亚锡-氯化钛还原重铬酸钾容量法(iso2597-1,iso2597-2、1so9507、gb/t6730.5、gb/t6730.65、gb/t6730.66),其缺点主要是铬和汞(iso2597-1:2006采用hgcl2氧化过量的sncl2)的污染问题。此外,测定铁矿中总铁含量时需要根据铁矿中含钒或铜或钼的含量确定采用何种前处理方法,给实际操作带来不便;同时,每次测定时必须采用有证标准物质进行验证,以确定所得结果是否正确,这无疑带来更大的工作量和成本的增加。其次,精度上也不尽人意,如gb6730.66-2009《铁矿石全铁含量的测定自动电位滴定法》,其精密度为sr=0.1,sr=0.21,即r=0.28%,r=0.59%(r=2.8sr,r=2.8sr)。其主要原因是采用重铬酸钾法测定铁,由于h+参与了反应,因此每次测定酸度必须保持一致,才能保证重复性限和再现性限较小,此外,目视确定滴定终点,带来了系统误差,特别是在预还原阶段,即用稀重铬酸钾氧化过量的还原剂时,误差更大。为了解决了上述问题,原广东检验检疫局提出《铁矿全铁含量的测定自动光度滴定法》(sn/t3100.1-2012)采用自动光度滴定法代替目视滴定法,用仪器自动判断滴定终点,消除了系统误差,方法中不涉及铬和汞的污染问题,是一种非常环保的测定方法。但该方法采用氧化还原滴定法,准确度仍有待提高。因此,开发一种前处理简单,检测结果准确度高的新的测定方法具有重要的研究意义和推广应用价值。技术实现要素:本发明的目的在于克服现有技术中铁含量的检测方法存在准确度不够,前处理复杂的缺陷或不足,提供一种铁矿石中全铁含量的测定方法。本发明提供的测定方法前处理操作简单,适用性强,测定结果准确度高,可应用于不同种类的铁矿含有不同范围的铁含量的测定。为实现上述发明目的,本发明采用如下技术方案:一种铁矿石中全铁含量的测定方法,包括如下步骤:s1:将铁矿石和固体碱置于刚玉坩埚中,加入过氧化钠混合均匀,于650~700℃下熔融20~30min得融结物,冷却;s2:向刚玉坩埚中加入水使得融结物溶解,加入盐酸、硝酸和硫酸溶液后加热至铁矿石溶解;所述融结物和盐酸的质量摩尔比为1:0.012~0.016g/mol;所述融结物和硝酸的质量摩尔比为1:0.018~0.020g/mol;所述融结物和硫酸的质量摩尔比为1:0.0045~0.0050g/mol。s3:再加入氟化铵和硼酸,搅拌条件下加入磺基水杨酸溶液和碱性溶液至ph为0.9~1.1;所述过氧化钠和氟化铵的质量比为1:0.3~0.4;所述氟化铵和硼酸的质量比为1:5~6;所述铁矿石和磺基水杨酸的质量比为1:1.8~2.2;s4:利用edta光度滴定法进行滴定,计算铁含量即可。铁矿石中钛含量较高,且edta滴定时,钛会对铁离子的滴定造成干扰,故需排除钛的干扰。另外,利用碱融法进行前处理时,坩埚的选取将影响铁含量的测定结果或影响其是否可测定。如选用常规的石墨坩埚,在熔融过程中会出现漏液的现象,如选用铁坩埚,将引入铁离子,影响最终的测定结果。经多次研究表明,选用刚玉坩埚,可实现铁矿石的较好熔融。但是选用刚玉坩埚来熔融时将会有al2o3析出,al2o3可与指示剂磺基水杨酸络合,会影响铁的滴定,故选用刚玉坩埚进行熔融时,后续需对铝进行掩蔽。本发明首先利用固体碱—过氧化钠对铁矿石进行熔融,然后利用盐酸和硝酸进行酸化溶解,然后利用硫酸络合钛生成硫酸钛,消除大部分钛的干扰,再加入氟化铵,一方面,利用f-与al3+络合,从而掩蔽大量的铝;另一方面,利用氟化铵与钛络合,进而避免钛的干扰;而氟化铵引入的氟离子也可与铁络合,使磺基水杨酸铁的溶度降低,吸光度降低,滴定终点突越不明显,故本发明选用硼酸来消除过量的氟化物,同时控制磺基水杨酸的加入量(比常规的加入量更高)使得滴定终点突越明显。本发明的测定方法前处理操作简单,适用性强,测定结果准确度高,可应用于不同种类的铁矿含有不同范围的铁含量的测定。优选地,s1中所述固体碱为氢氧化钠或氢氧化钾。更为优选地,s1中所述固体碱为氢氧化钠。优选地,s1中所述熔融的温度为650℃,时间为30min。优选地,s1中所述铁矿石和固体碱的质量比为1:7.7~8.3;所述铁矿石和过氧化钠的质量比为1:5.8~6.2。在上述条件下,可实现铁矿石的较好熔融。优选地,s1中所述固体碱为氢氧化钠;所述铁矿石和氢氧化钠的质量比为1:8;所述铁矿石和过氧化钠的质量比为1:6。优选地,s2中所述融结物和盐酸的质量摩尔比为1:0.014g/mol。优选地,s2中融结物和硝酸的质量摩尔比为1:0.019g/mol。优选地,s2中所述融结物和硫酸的质量摩尔比为1:0.0048g/mol。优选地,s3中加入碱性溶液至ph为1,所述碱性溶液为氢氧化钠溶液或氢氧化钾溶液。更为优选地,所述碱性溶液为氢氧化钠溶液。优选地,所述过氧化钠和氟化铵的质量比为1:2。熔融过程中,过氧化钠将会影响al2o3的析出量,通过调控过氧化钠和氟化铵的用量关系,可实现铝的较好屏蔽。在上述条件下,即可实现铝的较好屏蔽,由避免氟化铵的过量加入,及由其带来后续处理的成本增加。优选地,所述氟化铵和硼酸的质量比为1:6。优选地,所述铁矿石和磺基水杨酸的质量摩尔比为1:0.008g/mol。磺基水杨酸的过量加入可使得滴定终点突越明显,在该用量条件下,滴定终点突越明显,且不会造成过量浪费。本发明s4中的edta光度滴定法按常规的操作进行即可。与现有技术相比,本发明具有如下有益效果:本发明提供的测定方法前处理操作简单,适用性强,测定结果准确度高,可应用于不同种类的铁矿含有不同范围的铁含量的测定。附图说明图1为各种物质的吸收曲线;图2为滴定曲线(梅特勒公司自动电位滴定仪)(+字处为滴定终点);图3为滴定曲线(万通公司自动电位滴定仪)(bp2为滴定终点)。具体实施方式下面结合实施例进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下例实施例中未注明具体条件的实验方法,通常按照本领域常规条件或按照制造厂商建议的条件;所使用的原料、试剂等,如无特殊说明,均为可从常规市场等商业途径得到的原料和试剂。本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。铁矿中铁含量范围较宽,不同种类的铁矿含有不同范围的铁,如钒钛磁铁矿(原矿~精矿)13%~52%,磁铁矿fe3o4(fe:72.4%),赤铁矿α-fe2o3(fe:70.0%),菱铁矿feco3(fe:48.2%),褐铁矿fe2o3·nh2o(48~62.9%),针铁矿α-feo(oh)(fe:62.9%),镜铁矿(赤铁矿变种、与石英伴生)fe2o3(fe:70.0%)。进出口贸易的铁矿,铁含量大部分在50%~70%之间。因此,本方法应该选用有代表性的样品,基本可以覆盖所有含量范围。我们采用有证标准物质进行研究:表1有证标准物质证书数据单位:%各实施例中选用的试剂如下:高纯铁:纯度≥99.98%;氢氧化钠;过氧化钠(na2o2):无水;氟化铵(nh4f);硼酸(h3bo3);盐酸:ρ1.16g/ml;硝酸:ρ1.42g/ml;硫酸溶液:1+1;氢氧化钠溶液:200g/l;铁标准溶液:0.1000mol/l;乙二胺四乙酸二钠(c10h14n2na2o8,简称edta)标准滴定溶液:c(c10h14n2na2o8)=0.1mol/l;磺基水杨酸溶液:100g/l。其中,铁标准溶液通过如下过程配制得到:称取0.5585g铁粉放入250ml烧杯中,加入5ml盐酸和5ml硝酸。盖上表面皿,在80℃平板电炉上加热至完全溶解。冷却后转入100ml容量瓶中,用水稀释至刻度。乙二胺四乙酸二钠标准滴定溶液通过如下过程配制得到:称取40g乙二胺四乙酸二钠,加800ml水,加热溶解,冷却,稀释至1000ml,混匀。储存于塑料瓶中。标定的过程如下:(1)铁粉溶解称取0.20g(精确至0.0002g)铁粉放入250ml烧杯中,加入5ml盐酸和5ml硝酸。盖上表面皿,在80℃平板电炉上加热至完全溶解。在玻璃棒搅拌下,加入2ml硫酸溶液,15ml氢氧化钠溶液和5ml磺基水杨酸溶液,洗出玻璃棒,用水稀释至150ml。(2)滴定盖上表面皿,在210℃平板电炉上加热至沸(至没有小气泡)。取下烧杯,洗涤表面皿和烧杯内壁。趁热(溶液温度>80℃)将烧杯置于自动电位滴定仪滴定台上,插入复合玻璃电极,用电磁搅拌器子或仪器自带棒式搅拌器搅拌,在不断搅拌下,手动加入氢氧化钠溶液,调节溶液的酸度至ph(1±0.05)。插入光度电极和滴定头,确认光度电极和滴定头浸入溶液中,确保光度电极的光路没有气泡。运行滴定程序,在不断搅拌下,用edta标准滴定溶液滴定,滴定终点时消耗的edta标准滴定溶液体积记为v1。重复测定4次,记录4次的滴定终点v1,v2,v3,v4。在滴定过程中如发现有气泡,可暂停滴定,并将光度电极取出,离开溶液后再次插入溶液中,可消除气泡。注1:调酸度(ph)时不使用温度补偿。注2:滴定结束时溶液的温度必须保持在50℃以上。注3:万通自动搅拌设备搅拌速度设置为6(相当于溶液搅拌速度900r/min),梅特勒设备手动滴定台搅拌设备设置为40%(相当于控制搅拌子速度1520r/min,未知溶液转速与搅拌子大小/类型。注4:使用梅特勒dp5光度电极时,在调节溶液酸度至ph1后才插入溶液中,减少酸对光度电极的腐蚀。(3)计算未进行空白校准的edta浓度cn(u)按式(1)计算未进行空白校准的edta浓度cn(u)(n=1,2,3,4),以mol/l表示,结果计算到小数点后4位,式中:n1,2,3,4;mn纯铁粉的质量,g;vn滴定终点时消耗的edta标准滴定溶液的体积,ml;55.847铁的原子量,单位为克每摩尔,g/mol;1000体积的换算系数。如果四次结果(c1(u),c2(u),c3(u),c4(u))的极差(cmax-cmin)等于或小于0.0002mol/l,取四个结果的平均值作为最后结果cedta(u)。如果四次结果的极差大于0.0002mol/l,取中位值作为最后结果cedta(u)。4.11.2试剂空白移取1.00ml铁标准溶液(4.10),放入250ml烧杯中,加入5ml盐酸(4.6),5ml硝酸(4.7),2ml硫酸溶液(4.8)。在玻璃棒搅拌下,加入15ml氢氧化钠溶液(4.9)和5ml磺基水杨酸溶液(4.12),洗出玻璃棒,用水稀释至150ml。按程序4.11.2.2操作。记录滴定终点时消耗edta的体积(v01,v02,v03,v04)。如果四次结果(v01,v02,v03,v04)的极差(vmax-vmin)等于或小于0.02ml,取四个结果的平均值作为最后结果vb。如果四次结果的极差大于0.02ml,取中位值作为最后结果vb。按式(2)计算试剂空白值(v0),结果计算到小数点后2位。v0=vn-a(2)式中:a1.00ml铁标准溶液换算为等当量的edta标准滴定溶液的体积,ml。a按式(3)计算,结果计算到小数点后2位。式中:1.00铁标准溶液的体积,ml;0.1000铁标准溶液的浓度,mol/l;cedta(u)未进行空白校准的edta标准滴定溶液的浓度(4.11.3),mol/l。注5:如果溶液没有铁,不能形成磺基水杨酸铁络合物,无法进行光度滴定。因此进行空白试验时,需要加入铁,加入的铁量换算为等当量的edta标准滴定溶液的体积。注6:1ml单刻度移液管应先通过称量所移取的水的质量并换算成体积来进行校正。(4)计算edta标准滴定溶液的浓度cedta按式(4)计算edta标准滴定溶液的浓度,c0n(n=1,2,3,4),以mol/l表示,结果计算到小数点后4位,式中:n1,2,3,4;mn纯铁粉的质量,g;vn滴定终点时消耗的edta标准滴定溶液的体积,ml;v0试剂空白值(4.11.4),ml;55.847铁的原子量,单位为克每摩尔,g/mol;1000体积的换算系数。如果四次结果(c01,c02,c03,c04)的极差(cmax-cmin)等于或小于0.0002mol/l,取四个结果的平均值作为最后结果cedta。如果四次结果的极差大于0.0002mol/l,取中位值作为最后结果cedta。实施例1前处理最优条件探索本实施例对采用氢氧化钠-过氧化钠混合熔剂碱融法作为前处理方法。样品前处理条件试验采用正交试验法确认最优条件,包括:熔剂用量、熔融温度、熔融时间。样品量为0.25g。使用正交分析法来探索最佳的熔融条件,固定样品量为0.25g,主要因素有矿样水平、氢氧化钠用量、过氧化钠量、熔融时间、熔融温度4个因素。本实验是一个5水平试验,共有4个因素,不考虑因素间的交互作用,因此选用正交表l25(56)来试验。实验结果以回收率(检测结果与标准物质定值之比)作为参数。结果见表2和表3。表2实验正交表及实验结果表3实验数据方差分析表因素偏差平方和自由度f比f临界值显著性水平4.82240.4742.780nnaoh,g11.71941.1512.780nna2o2,g8.30140.8162.780n温度,℃13.04641.2822.780n时间,min13.04541.2822.780通过方差分析,熔融温度和熔融时间引起的误差最大,其次是氢氧化钠用量、过氧化钠用量。矿物种类影响最小。在所有条件下,f值都小于f临界值,说明所有条件均可用。直观判断,0.25g样品,氢氧化钠量1.5~2g,na2o2量1.5~2.5g,熔融温度600~700℃,熔融时间20~50min以上,分解效果最好。实施例2相关离子进行干扰试验edta与部分金属离子形成螯合物的形成常数见表4。表4edta与金属离子形成螯合物的形成常数金属离子fe3+bi3+co3+cr3+dy3+hf4+hg2+ho3+in3+logk25.127.840.7(23.4)18.329.521.718.625.0金属离子in3+ni2+pb2+sc3+sn2+th4+ti3+tl3+tm3+logk25.018.6218.0423.118.323.221.337.819.3从表4可以看出,四价的th4+、hf4+,三价的bi3+、in3+、co3+、cr3+、tl3+、稀土,在一定浓度下会产生影响,二价的hg2+、ni2+、pb2+、sn2+也可能会产生影响。但铁矿中上述离子的含量很低,见表5。表5铁矿中部分金属元素含量元素m铁矿中各元素的含量(%)fe3+46~72bi3+---hg2+---th4+---cu2+0.0004~1ni2+0.001~0.05pb2+0.001~0.05al3+0.06~5zn2+0.001~0.05cd2+---co2+0.0008~0.05稀土---我们选取bi3+、hg2+两个lgkm-edta大于或接近fe3+的元素和cu2+、al3+、co2+这三个铁矿中含量较大的元素,以及从稀土中选取la3+、eu3+、yb3+(分别代表轻、中、重稀土)进行干扰试验。cu2+由于与edta形成有色络合物,影响滴定终点的确认,因此,也有必要进行干扰试验,结果见表6。表6铁矿中部分金属元素含量(每组试验中加入的fe的量为55.846mg)试验结果表明,在本方法试验条件下,铁矿中各杂质元素对试验结果无明显影响。磺基水杨酸与金属离子配合物的稳定参数表见表7。表7磺基水杨酸与金属离子配合物的稳定参数表磺基水杨酸与过渡金属和重金属离子(特别是fe3+、uo2+以及al3+等金属)具有较高的络合稳定常数。在ph为1时,alf63-的logk=6.1,fef63-的logk=3.3,fe-edta的logk=7.3。但是,但由于采用碱熔法并用刚玉坩埚,会溶出大量的al2o3,必须对铝进行屏蔽,同时铝也会与磺基水杨酸络合,对铁的滴定有一定的影响,需要进行铝的干扰试验。本实施例利用nh4f使f-与al3+络合,从而掩蔽大量的铝,多余的f-与fe3+络合形成fef63,降低了溶液fe3+的浓度,使fe-ssa的浓度降低,吸光度减少,突跃平缓,为提高灵敏度,可增加ssa的浓度,如0.5gssa。h3bo3的加入可以络合edta取代fef63-的氟后产生的f-,进一步加快反应速度,缩短滴定时间本实施例选用添加不同配比的nh4f和硼酸来掩蔽铝,具体的实验条件如表8。表8铝的掩蔽效果表8显示了nh4f掩蔽铝的效果。从表8可以看出,0.5g的nh4f可以掩蔽0.15g的al(0.3gal2o3),也就是说,只要刚玉坩埚的损失量少于0.3g,在此条件下就可以完全掩蔽al。在实验的条件下:采用2gnaoh+1.5gna2o2作为熔剂,在650℃下熔融20min。刚玉坩埚的损失量小于0.05g(见表2),即熔出的al2o3<0.05g,,al<0.025g。因此,在本试验条件下,al的干扰可以消除。实施例3确定滴定条件(1)确定滴定的测定波长如图1,铁和铁与磺基水杨酸的络合物在400nm-700nm有吸收峰,edta、fe-edta络合物和滴定后的产物(fe-edta、磺基水杨酸)在500nm以上基本没有吸收。在520nm滴定时,铁与磺基水杨酸的吸收峰较高(不是最大),随着edta的加入,edta先与游离的铁络合生成络合物,络合物在此波长下没有吸收,溶液的吸光度a略变小(吸光度的主要贡献来自铁与磺基水杨酸的络合物),当游离铁被完全络合后,edta会取代铁与磺基水杨酸络合物中的磺基水杨酸,生成fe-edta和游离的磺基水杨酸,这些生产物在520nm下没有吸收峰,表现出吸光度快速变小,到达滴定终点和终点后,产物与过量的edta都不吸收,吸光度为0。由于滴定终点附近铁的浓度很小,反应慢,因此必须在较高温度下进行,使突变较明显,通过直线的拐点(两条直线外推),可以准确判断滴定终点。滴定曲线见图2和图3。edta与fe3+反应生产1:1络合物。在络合滴定中,为保持溶液的酸度和离子强度不变化,采用加入缓冲溶液的办法。edta滴定铁是在ph1强酸介质中进行,强酸本身就是缓冲溶液。(2)确定滴定终点用0.1mol/ledta在ph=1.0下滴定fe:a=abc……………………………………………………(1)cind为指示剂的总浓度。式(2)、式(3)相乘并合并:式(4)/式(5):滴定曲线任何一点:a=ai+ami整理式(7)和式(8)因此,等当点的吸光度为:由式(2)式(11)代入式(10)和为指示剂和指示剂络合物的最大吸光度,可从实验测出。如果已知道指示剂络合物的稳定常数kmi,就可以求出等当点时的吸光度。不过,在等当点时仍有微量的fe为指示剂络合,未完全与edta反应,必须按下列公式(13)加以校正。如果用极少量和足够灵敏的指示剂,则指示剂的校正可以忽略。如果kmi未测出,则可以认为拐点的吸光度ainfl作为等当点的吸光度aeq,则指示剂校正公式为:(3)理论计算用表8实际工作曲线的数据为例进行理论计算:吸2ml铁标准溶液(11.695mg,0.2094mol)用0.09444mol/ledta在ph1下滴定fe,测定波长520nm,以磺基水杨酸(分子量218.18)(10%)1ml为指示剂。开始滴定体积为150ml。cind=1×10%×1/218.18/150×1000=0.003056(mol/l)磺基水杨酸-fe络合系数:㏒k1=14.64㏒k2=10.54㏒k3=6.94。应文河用实验验证了在不同ph下,fe3+与磺基水杨酸逐级形成1-3型的络离子。在ph5形成1:2型fe3+-磺基水杨酸络离子(桔红色),在ph9~11.5形成1:3型fe3+-磺基水杨酸络离子(黄色)。在ph1形成的紫红色络离子应该为1:1型fe3+-磺基水杨酸。ph1时,logαy(h)=12.3。因此,㏒kmi=㏒k1-logαy(h)=14.64-12.3=2.3;edta--fe络合系数:㏒kfe-edta=25.1;edta酸效应系数:ph1,logαy(h)=18.01;edta-fe条件络合系数:ph1,logk’fe-y=25.1-18.01=7.09。在ph1,edta-fe条件络合系数大于1:1型fe3+-磺基水杨酸的条件络合系数,可以取代1:1型fe3+-磺基水杨酸络合物的铁。以仪器给出的终点:2.141ml,a=0.05。此时fe的浓度为(mol/l):(假如溶液体积为150ml)cfe=11.695/1000/55.847/0.15=0.0014等当点时,[m]eq=cfekfe-edta=0.0014107.09=1×10-5[m]eqkmi=10-5×102.3=0.002aeq=aimax+amimax[m]eqkmi1+[m]kmi=0.048+2.047×0.0021+0.002=0.052查aeq=0.052时,edta消耗体积为:2.141ml。进行指示剂校正:mieq=aeq-aimaxamimax-aimaxcind=0.052-0.0482.047-0.048×0.003=6×10-6以等当点时的体积为152ml则校正系数相当于0.09444mol/ledta毫升数:6×10-6×152/0.09444=0.01因此可以认为滴定终点为2.141+0.01=2.151ml。理论计算值与仪器给出的数据一致。如果标定时也采用相同的判断终点的办法,可以认为仪器给出的滴定终点就是实际的滴定终点。表9实际滴定曲线数据(edta滴定fe)滴定条件主要考虑溶液的ph值、温度、指示剂量、滴定模式和滴定终点的判断。由于edta滴定为非常成熟的滴定方法,故除指示剂用量外,其他参数采用《gb/6730.73-2016铁矿石全铁含量的测定edta光度滴定法》的结果,即加热溶液至近沸,在ph为1下进行滴定,采用动态加入法,用折线模式判断滴定终点。(4)指示剂用量试验在前述最佳前处理条件下,需要0.5gnh4f掩蔽刚玉坩埚损耗的al,氟会与铁络合,降低了指示剂磺基水杨酸(简称“ssa”)与铁的络合能力,为使滴定终点突越变得更明显,必须加入硼酸掩蔽氟,同时增加指示剂ssa的用量。如表9,为实验条件和结果。表10ssa用量对结果的影响试验编号fe,mgnh4f,gh3bo3,gssa,g开始时吸光度aedta,ml111.695000.12.1172.16211.695000.52.1902.15311.6950.530.51.4392.16411.6951311.7632.15表10表明,0.1~1g的指示剂对结果没有影响。随着nh4f加入量的增加,开始时吸光度逐步下降,表明即使加入3g硼酸,部分铁仍与氟络合,降低了铁与指示剂ssa的络合物浓度,吸光度变小。但在该滴定条件下,滴定结果不变。实施例4方法学验证对最佳的测定方法进行方法学验证。具体的测定方法为:将0.25mg试样置于盛有2.5g氢氧化钠的刚玉坩埚中,再加入1.5g过氧化钠,混匀。将坩埚置于650℃的高温炉中熔融30min。从高温炉中取出坩埚,冷却。将坩埚置于盛有250ml烧杯中,加入100ml温水,用玻璃棒搅拌坩埚使融结物溶解,加入2ml硫酸溶液,5ml盐酸和5ml硝酸,加入1-2颗玻璃珠(防止加热爆沸),盖上表面皿,缓缓加热至试料溶解。用水洗出坩埚。加入0.5g氟化铵和3g硼酸。在玻璃棒搅拌下,加入5ml磺基水杨酸溶液和5ml氢氧化钠溶液,洗出玻璃棒,用水稀释至150ml。(1)方法的线性范围从方法的线性范围可以推断出标准的测定范围,当然,为获得精密度数据,标准的测定范围由精密度所用的各水平确定。(2)方法正确性的验证采用有证标准物质对方法进行验证,确保方法的正确性。本试验验证样品7个,覆盖整个检测范围。具体的测定结果如下:使用有证标准物质验证方法的准确度。结果见表11。比对评价程序:设方法测得的结果为x,标准物质的定值为x0。ux,ux0分别为它们的不确定度(k=2),判断测定结果与定值是否存在差异的依据为:准则:en≤1,表明测量结果质量是有保证的。拒绝准则:en>1,表明测量结果质量可能得不到保证。从表11可以看出,本方法的en<1,结果是可靠的。方法与gb/t6730.5比较,使用实际进出口样品进行试验,结果见表12。表11标准样品的测试结果表12实际进出口样品检测结果与gb/t6730.5-2007方法的结果基本上差值<0.3%,方法准确可靠。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1