一种药品中钯金属残留的测定方法与流程
本发明属于重金属残留物检测技术领域,尤其涉及一种药品中钯金属残留的测定方法。
背景技术:
化学药合成过程中的起始原料、催化剂、副产物、未反应完全的试剂、金属容器、管道以及其他不耐酸碱的金属工具均可能引入金属杂质,而原辅料中的金属残留物会进一步带入到药物制剂中。钯在有机合成中是一种常用的催化剂,在药品生产中也经常使用,但钯是一种重金属,对人体有一定的危害。
药品生产中残留的钯存在的形态不确定,可能是金属钯、氧化钯或化合物钯,对样品的处理增加了难度。目前,常用的钯残留检测方法是原子吸收分光光度法,样品处理的方法有硝酸-高氯酸消解法、王水直接溶解法及600度灰化王水溶解法等。上述单一的样品处理方法很难满足实验要求,况且金属钯是一种非常好的催化剂,在样品的处理过程中易转变为氧化钯。金属钯易溶于王水和热的硫酸及浓硝酸,但是氧化钯不溶于王水,所以采用硝酸-高氯酸消解法、王水直接溶解法处理试样仅能测试样品中的金属钯及钯盐的含量;采用样品600度灰化,加王水溶解,然后再加盐酸赶除硝酸方法处理试样,在600度灰化时有一部分金属钯转变为了氧化钯,造成测定结果偏低。
技术实现要素:
本发明的目的是克服上述现有钯残留测定方法存在的缺陷,提供一种能够提高药品中钯残留测定准确度和精密度的方法,该方法能准确的测定出药品化合物中的钯含量,且用酸量少,减少了对环境的污染。
为了达到上述目的,本发明采用以下技术方案。
一种药品中钯金属残留的测定方法,包括以下步骤:
⑴标准曲线溶液配制
100μg/ml钯溶液:精密吸取5.0ml、1000μg/ml钯标准溶液于50ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
0.0μg/ml钯溶液:2%的盐酸溶液;
0.5μg/ml钯溶液:精密吸取0.5ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
1.0μg/ml钯溶液:精密吸取1.0ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
2.0μg/ml钯溶液:精密吸取2.0ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
4.0μg/ml钯溶液:精密吸取4.0ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
⑵绘制标准曲线:使用岛津aa-6650原子吸收分光光度计,在波长:247.6nm、电流:10ma、空气/乙炔焰:1.6~1.8:1条件下,进样器分别取0.0μg/ml、0.5μg/ml钯溶液、1.0μg/ml钯溶液、2.0μg/ml钯溶液、4.0μg/ml钯溶液,依次测定吸光度,然后以吸光度为纵坐标,浓度为横坐标,绘制标准曲线,得到标准曲线公式;
⑶制备供试样品溶液
精密称取适量供试样品加入坩埚中,加入1ml分析纯硫酸,在电炉上完全碳化,转入580°~620°的高温炉中完全灰化,取出加入5ml水与1ml甲酸在电炉上小火蒸至近干,后加入3ml王水,在电炉上小火蒸至近干,再每次加入2ml分析纯盐酸在电炉上赶硝酸,重复3次,转移至10ml容量瓶中,用水稀释至刻度,混匀;
⑷制备空白溶液
取1ml分析纯盐酸加入坩埚中,加入1ml分析纯硫酸,在电炉上完全碳化,转入580°~620°的高温炉中完全灰化,取出加入5ml水与1ml甲酸在电炉上小火蒸至近干,后加入3ml王水,在电炉上小火蒸至近干,再每次加入2ml分析纯盐酸在电炉上赶硝酸,重复3次,转移至10ml容量瓶中,用水稀释至刻度,混匀;
⑸浓度测定:在标准曲线的同等仪器条件下,测定供试样品溶液的吸光度,从标准曲线上求得供试样品溶液的浓度ρ1;在标准曲线的同等仪器条件下,测定空白溶液的吸光度,从标准曲线上求得空白溶液的浓度ρ0;
⑹计算:供试样品中钯金属残留量为
其中:ρ1—表示供试样品溶液代入标准曲线公式得到的浓度,单位:μg/ml;
ρ0—表示空白溶液代入标准曲线公式得到的浓度,单位:μg/ml;
v—表示供试样品定容体积,单位:ml;
m—表示供试样品质量,单位:g;
完成药品中钯金属残留的测定。
上述2%盐酸溶液是吸取20ml分析纯盐酸,用水稀释至1000ml得到的溶液;
上述王水是取300ml分析纯盐酸,向其中缓慢加入100ml分析纯硝酸,搅拌并混合均匀得到的溶液;
上述步骤⑶和⑷中采用的坩埚为玻璃或石英制品。
上述步骤⑶和⑷中,碳化时电炉先小火碳化再逐渐升高电炉温度至完全碳化。
与现有技术相比,本发明的有益技术效果:
1、将供试样品先高温灰化,有机化合物中钯转化为金属钯或氧化钯,加入甲酸把氧化钯还原为金属钯,蒸至近干,再加入王水溶解金属钯,提高了药品中钯金属残留测定结果的精密度和准确度;
2、由于大量硝酸会影响原子分光光度分析,本发明在标准曲线溶液配制及供试样品制备时采用分析纯盐酸,并利用盐酸精准加热控制赶出硝酸,降低了溶液中的硝酸浓度,降低了硝酸对原子分光光度分析的影响。
3、选用玻璃或石英坩埚,避免使用白色瓷坩埚对供试样品进行灼烧处理,保证测定结果的精密度和准确度;碳化时电炉先小火碳化再逐渐升高电炉温度至完全碳化,防止恒温碳化易造成供试样品外溢从而影响测定结果的精密度和准确度。
附图说明
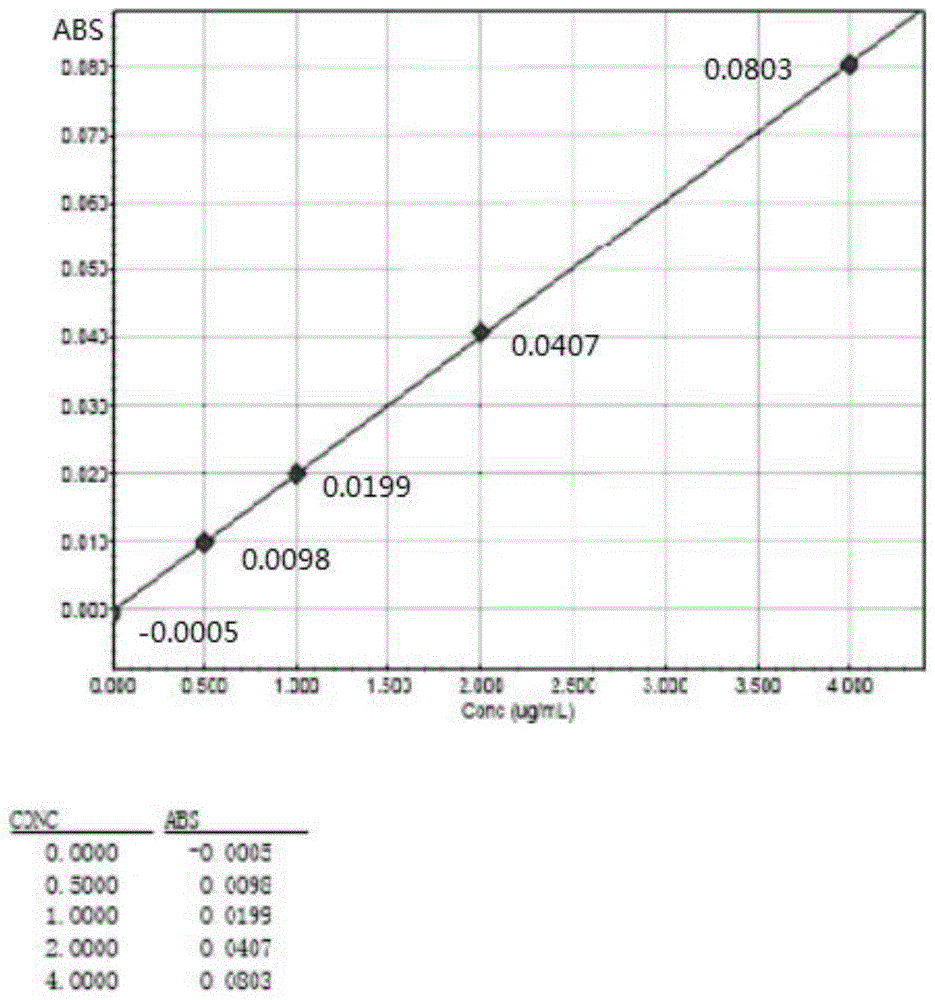
图1为本发明的药品中钯金属残留的测定方法标准曲线图。
具体实施方式
下面将结合本发明具体实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
实施例
1、仪器与试剂
1)原子吸收分光光度计型号aa-6650,钯灯247.6nm,电流10ma,空气/乙炔焰:1.6~1.8:1,
2)钯标准溶液1000ug/ml
3)硫酸、硝酸、盐酸、甲酸、高氯酸均为分析纯级试剂。
4)2%盐酸溶液:吸取20ml分析纯盐酸,用水稀释至1000ml;
5)王水:取300ml分析纯盐酸,向其中缓慢加入100ml分析纯硝酸,搅拌并混合均匀;
2、标准曲线溶液配制
100μg/ml钯溶液:精密吸取5.0ml、1000μg/ml钯标准溶液于50ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
0.0μg/ml钯溶液:2%的盐酸溶液;
0.5μg/ml钯溶液:精密吸取0.5ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
1.0μg/ml钯溶液:精密吸取1.0ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
2.0μg/ml钯溶液:精密吸取2.0ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
4.0μg/ml钯溶液:精密吸取4.0ml、100μg/ml钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
3、绘制标准曲线:使用岛津aa-6650原子吸收分光光度计,在波长:247.6nm、电流:10ma、空气/乙炔焰:1.6~1.8:1条件下,进样器分别取0.0μg/ml、0.5μg/ml钯溶液、1.0μg/ml钯溶液、2.0μg/ml钯溶液、4.0μg/ml钯溶液,依次测定吸光度,然后以吸光度为纵坐标,浓度为横坐标,绘制标准曲线,如图1所示,
标准曲线公式为:abs=0.02021ρ+0.0000275001,r=0.9999,ρ为浓度;
4、制备供试样品溶液
精密称取适量供试样品加入坩埚中,加入1ml分析纯硫酸,在电炉上完全碳化,转入580°~620°的高温炉中完全灰化,取出加入5ml水与1ml甲酸在电炉上小火蒸至近干,后加入3ml王水,在电炉上小火蒸至近干,再每次加入2ml分析纯盐酸在电炉上赶硝酸,重复3次,转移至10ml容量瓶中,用水稀释至刻度,混匀;
5、制备空白溶液
取1ml分析纯盐酸加入坩埚中,加入1ml分析纯硫酸,在电炉上完全碳化,转入580°~620°的高温炉中完全灰化,取出加入5ml水与1ml甲酸在电炉上小火蒸至近干,后加入3ml王水,在电炉上小火蒸至近干,再每次加入2ml分析纯盐酸在电炉上赶硝酸,重复3次,转移至10ml容量瓶中,用水稀释至刻度,混匀;
6、浓度测定:在标准曲线的同等仪器条件下,测定供试样品溶液的吸光度,从标准曲线上求得供试样品溶液的浓度ρ1;在标准曲线的同等仪器条件下,测定空白溶液的吸光度,从标准曲线上求得空白溶液的浓度ρ0;
7、计算:供试样品中钯金属残留量为
其中:ρ1—表示供试样品溶液代入标准曲线公式得到的浓度,单位:μg/ml;
ρ0—表示空白溶液代入标准曲线公式得到的浓度,单位:μg/ml;
v—表示供试样品定容体积,单位:ml;
m—表示供试样品质量,单位:g;
完成药品中钯金属残留的测定。
上述供试样品采用的坩埚为玻璃或石英制品;若供试样品在瓷坩埚中灼烧处理,白色的坩埚使用后底部发黄,疑是钯的化合物,选用石英坩埚则没有此现象。
上述碳化时电炉先小火碳化再逐渐升高电炉温度至完全碳化,供试样品碳化时未从坩埚中溢出。
试验例
选取三个批次的艾曲波帕为试验样品。
1)艾曲波帕样品的钯含量检测
三批次艾曲波帕中各批次样品随机各抽取2个样品,按实施例的方法进行检测,检测结果如表1所示。
表1三批次艾曲波帕样品的钯含量
2)精密度测试
10ug/ml钯溶液配制:精密吸取10.0ml、100ug/ml的钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
在第一批次中随机抽取6个艾曲波帕样品,进行编号,按编号分次精密称取艾曲波帕2g加入2ml、10ug/ml钯溶液,按实施例中制备供试样品溶液的处理方法进行处理,同时进行空白试验,并记录试验结果,表1为精密度测定的试验数据。
表2精密度测试数据(浓度:定容10ml溶液中钯的浓度)
3)准确度测试
10ug/ml钯溶液:精密吸取10.0ml、100ug/ml的钯溶液于100ml容量瓶中,用2%盐酸溶液定容至标线,混匀;
50%回收率测试
在第一批次中随机抽取3个艾曲波帕样品,进行编号,按编号分次精密称取艾曲波帕2g加入1ml、10ug/ml钯溶液,按实施例中制备供试样品溶液的处理方法进行处理;
100%回收率测试
在第一批次中再次随机抽取3个艾曲波帕样品,进行编号,按编号分次精密称取艾曲波帕2g加入2ml、10ug/ml钯溶液,按实施例中制备供试样品溶液的处理方法进行处理;
150%回收率测试
在第一批次中再次随机抽取3个艾曲波帕样品,进行编号,按编号分次精密称取艾曲波帕2g加入3ml、10ug/ml钯溶液,按实施例中制备供试样品溶液的处理方法进行处理;
同时进行以上艾曲波帕样品的钯含量测试及空白试验,并记录试验结果,表2为准确度测定的试验数据。
表3准确度测试数据如下(浓度:定容10ml溶液中钯的浓度)
通过上述试验例数据表明,本发明的方法准确度和精密度较高,且测定出的是有机物中的总钯含量,能准确的反应出有机化合物中钯含量。此方法使用酸量少,减少了对环境的污染。
基于以上内容可知,本发明的方法具有应用于所有有机物中微量钯含量检测的价值。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!