用于鉴别当归与混伪品东当归的方法与流程
本发明涉及一种用于鉴别当归与混伪品东当归的方法,具体涉及一种基于色谱-制皮联用的当归鉴别方法,属于中药质量检测技术领域。
背景技术:
当归为伞形科二年生草本植物当归angelicasinensis(oliv.)diels的干燥根,药用记载始见于《神农本草经》,距今已有2000多年历史。当归有浓郁的香气,味甘、辛、微苦,具补血活血,调经止痛,润肠通便之功效。主要用于血虚萎黄,眩晕心悸,月经不调,经闭痛经,虚寒腹痛,风湿痹痛,跌扑损伤,痈疽疮疡,肠燥便秘等症。在1983年资源普查之后,国家医药管理部门明确规定,禁止以欧当归、东当归等代替当归入药,《中国药典》2005版收载的当归来源仅为伞形科植物当归干燥根。由此可见,当归植物基源非常明确,且是单一植物基源,为我国特有种,即当归基源植物为伞形科植物当归[angelicasinensis(oliv.)diels]。
东当归(a.acutilobakitag)是当归的主要混伪品。东当归为当归属植物东当归angelicaacutiloba(sieb.etzucc)kigusticumacutilobum.siebetzuce的根,别名日本当归、大和当归、延边当归。现有技术中多采用性状、显微和理化鉴别等方法对当归和东当归进行鉴别,例如,现有公开的科技文献:王翰华,胡双丰.当归及其常见伪品东当归和欧当归的鉴别[j].中国医药指南,2012.10(5):4-5。另,还有基于its2条形码能够有效地鉴别中药当归及其混伪品,目前多采用pcr技术,从分子生物学水平上利用核糖体rrna基因内转录间隔区(its2)序列,譬如,现有公开的科技文献:张春,王晓丽,朱烨等.中药当归及其混伪品的rdnaits序列分析与鉴别[j].四川农业大学学报,2011.29(2):218-224。然而,上述现有当归鉴别技术,均存在一定的现实缺陷,其中,传统的鉴别技术受原植物生长环境、生长期及炮制加工工艺的影响;而,dna条码技术又受加工、炮制等影响dna模板质量工序的影响。因此,传统的鉴别技术和分子生物学技术,存在难以稳定、可靠、快速的实现该药材的真伪鉴定。
色谱-质谱联用鉴定技术是一种将鉴别、定量性能高效结合的技术。因此,本发明技术方案,其是基于色谱的高效分离性能与质谱技术所获取的丰富的成分结构信息,二者的有机结合便可促进系统中药鉴定技术的形成,有利于促进传统检测器选择性与灵敏度的显著提高,还可以定性中药样本中的未知化学成分。并结合多元统计分析技术,不但能提高不同来源、产地药材的辨别能力,更适用于加工、炮制后极易出现变化与损坏的中药样品。为此,本发明经过反复摸索和验证,创造性的建立一种快速、准确鉴别当归及其伪品东当归的方法。
技术实现要素:
本发明的目的在于提供一种基于主成分分析的当归及其混淆品东当归的色谱-质谱联用鉴别方法,最终以当归中藁本内酯:蛇床子素的s/n比值(s/n藁本内酯:s/n蛇床子素)作为鉴别其与混伪品的鉴别指标。
为了达到上述目的,本发明采用以下技术方案予以实现:
一种用于鉴别当归与混伪品东当归的方法,所述的当归是当归药材或饮片;所述的鉴别方法,是以当归中藁本内酯:蛇床子素的s/n比值作为鉴别其与东当归的检测指标。本发明方案中所述的s/n比值,亦即为色谱峰的信号/噪音比值。
进一步地,本发明所述的当归与混伪品东当归的鉴别方法,其特征在于:所述测定指标,其当归中藁本内酯:蛇床子素的s/n比值大于1,东当归中藁本内酯:蛇床子素的s/n比值小于1。
再进一步,本发明所述的用于鉴别当归与混伪品东当归的方法,所述的鉴别方法是采用色谱与质谱相联用的技术而建立。
再进一步,本发明所述的当归与混伪品东当归的鉴别方法,所述的鉴别方法中色谱条件如下:采用c18色谱柱;流动相为0.1%甲酸-水溶液与乙腈的体积比;流速为300μl/min,柱温为30℃,进样量为5μl,检测波长254nm,梯度洗脱参数:洗脱时间0-1min,0.1%甲酸-水溶液体积占比为98%;洗脱时间1-18min,0.1%甲酸-水溶液体积占比从98%到0%;洗脱时间18-20min,0.1%甲酸-水溶液体积占比为0%;洗脱时间20-23min,0.1%甲酸-水溶液体积占比从0%-98%;洗脱时间23-25min,0.1%甲酸-水溶液体积占比为98%。
更进一步,本发明所述的当归与混伪品东当归的鉴别方法,所述的鉴别方法的质谱条件如下:离子源为电喷雾离子源,正负离子模式采集数据,正负离子源喷射电压分别为5500v、-4500v,雾化温度550℃,雾化气和辅助气为氮气,均为50psi,气帘气35psi;母离子扫描范围50-1600m/z,对超过100cps的8个最强峰进行ms2采集数据,子离子扫描范围为50-1500m/z。
进一步,本发明所述的当归与混伪品东当归的鉴别方法,所述的鉴别方法中当归或东当归样品溶液制备方法如下:称取当归或东当归粉末0.2g,精密称定,置于锥形瓶中,精密加入20ml70%甲醇,称定重量,85℃水浴回流30分钟,静置放冷,称定重量,加入溶剂补足减少的重量,摇晃均匀后静置,取上清液过滤,取续滤液,即得。
另,本发明的技术方案中,对于液相色谱条件进行了优化,具体如下:
流动相分别比较了乙腈-水和甲醇-水两个溶剂系统,结果表明,用乙腈-水二元梯度洗脱系统比用甲醇-水梯度洗脱基线平,且耗时短。分析时间仅用30min,样品1h谱图显示,30分钟后无特征峰出现。以∑rs,r*(×10-10),ф,hcrf评价样品的分离效能:
hcrf=1,000,000n+100,00rmin+(tm-t1)(公式3)*:分层色谱响应值
*r:两峰间分离度,n:理论塔板数,rmin:最小分离度,tm-t1:最大与最小保留时间之差,a:色谱峰面积。
∑rs是指基于uspresolution相邻两峰的分辨率之和,主要用于评价色谱分离质量。由于∑rs通常由具有最大分辨率的谱峰决定,而最大分辨率往往与指纹图谱分离的总效能值相关较小。因此,使用r*(×10-5)(公式1)值可以使指纹图谱中所有谱峰的分辨率保持在归一化的状态。另一个用于评价指纹图谱分离质量的参数是ф值(公式2),ф值也叫色谱信息量值,是基于色谱峰面积、保留时间与理论塔板数来建立指纹图谱的色谱交互信息值。ф值有助于分析时间较短,药物活性成分较多的指纹图谱分析。本标准中还采用了分层色谱响应值(hcrf,公式3)对方法中分离峰个数,分离度和分析时间性能作出评价。hcrf值能对分离度,分离时间较差的指纹图谱作出合理的评价。因此,为了从多方面评价指纹图谱的质量,本发明方案中采用以上四个响应因子对色谱质量评价。
为了获得最佳的参数设置,选择柱温(25-35℃),流速(0.2-0.5ml/min),缓冲液浓度(0.1-0.2%甲酸,0.05-0.1%乙酸),水相初始比例(95-99%),色谱柱类型(柱1:50mm×2.1mm,1.7μm;柱2:100mm×2.1mm,1.7μm;柱3::100mm×2.1mm,2.5μm)作为优化参数,结果见表1。
表1色谱条件优化结果
优化结果显示,缓冲系统为0.1%甲酸-水溶液,柱温为30℃时,水相初始比例为98%,流速为0.3ml/min时,各峰分离度,塔板数参数优化结果较好,因此将此作为确定的色谱条件进行样品测定。
本发明的有益效果
1.本发明技术方案,建立了联用色谱-质谱技术鉴别当归与混伪品东当归的检测指标,亦即藁本内酯:蛇床子素的s/n比值,具有准确、快速的技术效果,明显克服了现实中传统鉴别方法与分子生物学的技术缺陷。
2.本发明所述的技术方案,充分遵从中医药成分到效果的综合评价理念,摈弃了现有技术中为单一成分而定的片面方式,进而有效的确保中药当归的用药安全和疗效质量。
附图说明
图1是本发明一个实施方案的当归和东当归正离子模式总离子流图;
图2是本发明一个实施方案的当归和东当归pca拟合散点图;
图3是本发明一个实施方案的当归和东当归成分差异载荷图;
图4是本发明一个实施方案的当归和东当归最大差异离子比较图;
图5是本发明一个实施方案的蛇床子素色谱图;
图6是本发明一个实施方案的蛇床子素一级质谱图;
图7是本发明一个实施方案的蛇床子素二级质谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
实施例一种当归和混伪品东当归的鉴别方法
(1)样品溶液的制备
1)当归药材溶液的制备:称取当归粉末(过三号筛)约0.2g,精密称定,置于锥形瓶中,精密加入20ml70%甲醇,称定重量,85℃水浴回流30分钟,静置放冷,称定重量,加入溶剂补足减少的重量,摇晃均匀后静置,取上清液过滤,取续滤液备用。
2)东当归溶液制备:称取东当归粉末(过三号筛)约0.2g,精密称定,置于锥形瓶中,精密加入20ml70%甲醇,称定重量,85℃水浴回流30分钟,静置放冷,称定重量,加入溶剂补足减少的重量,摇晃均匀后静置,取上清液过滤,取续滤液备用。
(2)uplc-q-tof/ms定性分析
1)质谱条件:离子源为电喷雾离子源,正负离子模式采集数据,正负离子源喷射电压分别为5500v、-4500v,雾化温度550℃,雾化气和辅助气为氮气,均为50psi,气帘气35psi;采用信息依赖采集(ida),动态背景扣除(dbs)和高灵敏度模式采集数据,母离子(tof-ms)扫描范围50~1600m/z,对超过100cps的8个最强峰进行ms2采集数据,子离子扫描范围为50~1500m/z。
2)液相方法:采用acquityuplcbehc18柱(50mm×2.1mm,1.7μm)色谱柱;流动相为0.1%甲酸-水溶液a与乙腈b;流速为300μl/min,柱温为30℃,进样量为5μl,检测波长254nm;梯度洗脱条件,如表2所示。
表2梯度洗脱条件
3)特征检测离子的确定
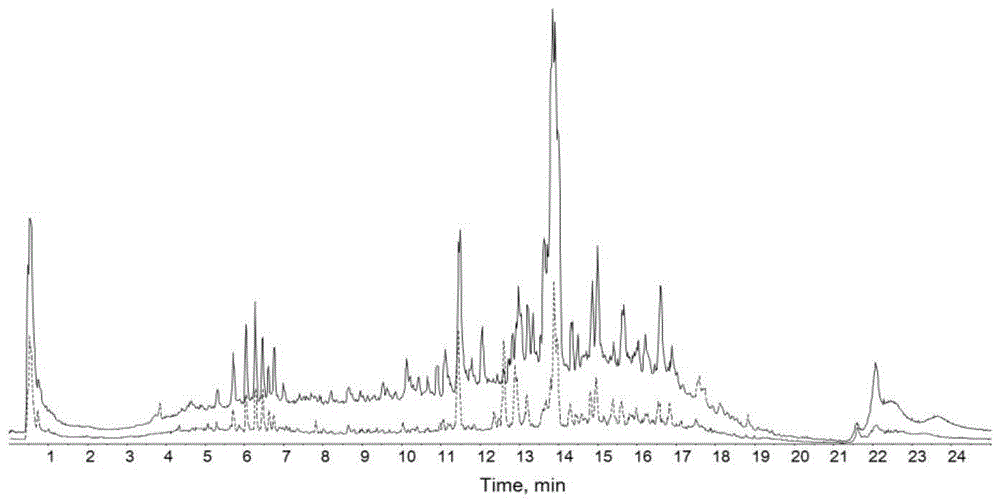
tripletoftm5600+可以提供化合物的精确质量数和同位素峰度比,以peakview2.2查看采集数据,以masterview在数据库中进行非靶向模式成分搜索,参数设置如下:设定masserror(质量数偏差)<5.0,权重30%;isotope(同位素分布差异)<10.0%,权重30%;fomularfinderscore(分子式查找)>70,权重40%。以精确提取化合物的准分子离子,质量偏差、保留时间和二级质谱图,确定化合物信息。当归和东当归正离子模式总离子流图见如图1所示(实线-当归;虚线-东当归)。
将ida采集的数据与中药成分数据库进行化合物匹配,并将数据导入markerview1.3进行主成分分析(principalcomponentanalysis,pca),找到特征性差异成分。经标度化(autoscaling)预处理数据后,建立正负离子模式数据的pca模型,从药材分类的主成分散点图可以看出,在正离子模式下,东当归采样样本点显示出与其他批次当归正品药材较大的差异,具体见图2所示。根据生成的分类结果,提取组间差别较大的化合物信息,筛选对聚类差异贡献较大的化合物离子,筛选规则为倍数差异(foldchange,fc)>1.5,t检验结果p<0.01,结果见图3。根据差异化合物质荷比和保留时间,匹配absciex(美国应用生物系统公司)中药化合物质谱数据库检索精确质量质谱信息,找到重要差异成分蛇床子素(质荷比245.1m/z,保留时间11.4min),当归和东当归中蛇床子素成分差异,具体结果见图4所示。
根据上述实验结果,配置蛇床子素0.01mg/ml的对照品溶液,按照上述进样条件进样,蛇床子素保留时间为11.440min,结果见图5。根据一级质谱结果,选择响应好且相对稳定的离子对作为检测指标,采用超高效液相色谱串联三重四级杆质谱法对各离子的碰撞能量进行了优化,结果见图6。其中,特征正离子为m/z245.1→189.0,m/z245.1→131.0,ce:22.8,dp:79.6;cxp:6.15,结果见图7。
(3)当归与东当归鉴别方法的确定
1)专属性考察:以不含当归的溶液为阴性样品,以东当归对照药材和待检样品为阳性样品,选择特征离子峰m/z245.1→189.0,m/z245.1→131.0作为检测离子对进行测定,结果阴性样品中的色谱图中,在与对照药材及样品相应的位置上无相应色谱峰,说明方法的专属性良好。
2)检测限考察:按照上述(1)中所述的样品溶液制备当归和东当归鉴别溶液,以主要成分藁本内酯(定量参数:m/z191.0→115.0)作为参考峰,考察当归和东当归中蛇床子素和藁本内酯色谱峰的信号/噪音(s/n)比,结果如表3所示。
表3当归藁本内酯:蛇床子素的s/n比值(s/n藁本内酯:s/n蛇床子素)结果
结果显示:东当归藁本内酯:蛇床子素的s/n比值(s/n藁本内酯:s/n蛇床子素)小于1,而15批当归藁本内酯:蛇床子素的s/n比值大于1。因此,在当归药材色谱中,其藁本内酯(c12h14o2)与蛇床子素(c15h16o3)的信号峰比值不得小于1,可作为当归和东当归鉴别的检验标准。
- 还没有人留言评论。精彩留言会获得点赞!