区分同量异序氨基酸和氨基酸组合的标记方法与流程
1.本发明涉及多肽测序方法。具体地,本发明涉及在多肽测序期间区分同量异序氨基酸和氨基酸组合:天门冬酰胺和甘氨酸
‑
甘氨酸;谷氨酰胺和甘氨酸
‑
丙氨酸;和/或谷氨酰胺和丙氨酸
‑
甘氨酸的方法。
背景技术:
2.对于肽测序,质谱已使用了数十年。通常,将给定的肽分离并碎片化。在低碰撞能下,在肽键或其附近发生肽碎片化。基于碎片化模式的准确位置,将碎片离子,包括肽的n末端命名为a、b或c离子,并且将它们来自c末端的各个互补片段分别命名为x、y或z。y1离子将表示c末端侧上的第一个氨基酸残基,而b1将表示肽n末端侧上的第一个氨基酸残基;y3离子将是由肽c末端侧上的最后3个氨基酸组成的离子并且与b离子b(n
‑
3)互补,其中“n”是特定肽中的氨基酸残基数目。使用ethcd(电子传递解离高能碰撞诱导解离)使得能够产生与侧链切割有关的其他类型的离子(d和w离子)(wysocki等人2005;johnson等人1988;frese等人2012)。
3.胰蛋白酶是将蛋白切割成短肽的蛋白酶,其通常更易于通过质谱分析和测序。使用胰蛋白酶而不是任何其他蛋白酶的优势在于由胰蛋白酶作用所产生的肽的两端通常具有正电荷,因此一旦ms/ms碎片化,两端碎片通常是通过质谱可检测的。在酸性条件(对于ms分析,通常在ph 2至4之间)下,肽的n末端质子化(pka 8.2)并且类似地,c末端也从k(赖氨酸,pka 9.74)或r(精氨酸,pka 10.76)侧链质子化,它们是胰蛋白酶的2个切割位点。使用胃蛋白酶水解,n末端碎片离子将是可检测的,但是对于c末端碎片,仅包括k或r的碎片离子将很可能是可检测的。由除胰蛋白酶或lys
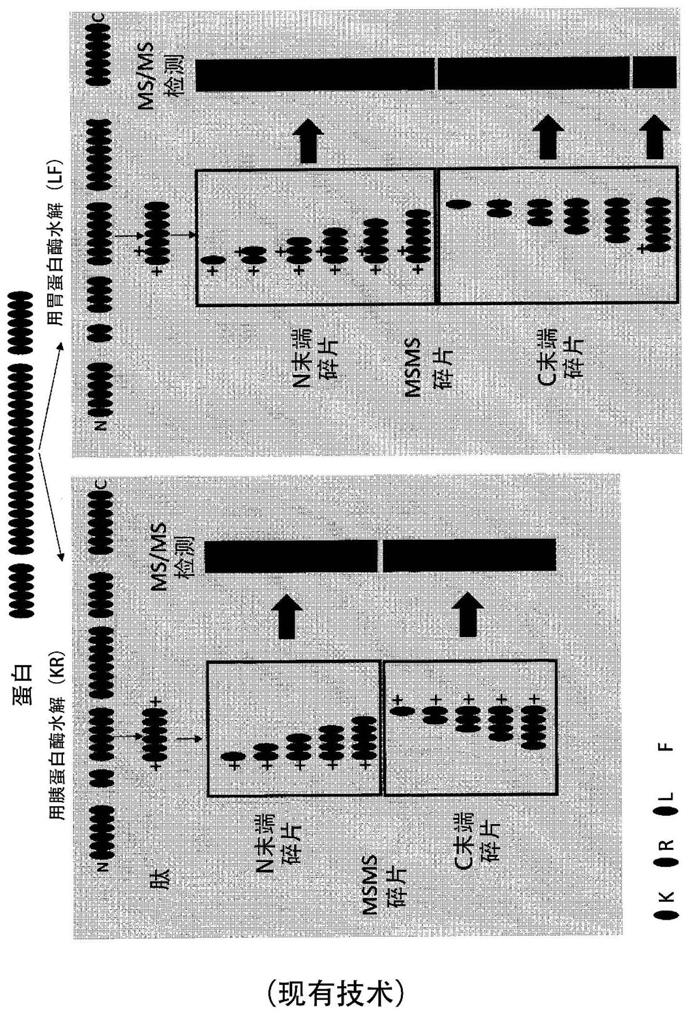
‑
c以外的蛋白酶所产生的c末端碎片通常由于它们不良的电离状态而不能被检测。图1显示了从胰蛋白酶相对于胃蛋白酶的作用所产生的蛋白切割,以及可以在ms/ms测序中使用的碎片。绿色的柱显示了应电离,因此通过质谱仪可检测的肽。与所使用的蛋白酶无关,大部分来自n末端肽的碎片应电离(通过每种水解物右侧的绿色柱显示)。另一方面,如果它们含有碱性残基之一(k、r或h),则来自肽的c末端的肽碎片仅将被检测。图1右侧的红色柱显示了不含任何碱性残基并因此通常通过质谱仪不可检测的c末端肽碎片。
4.从质/荷肽谱的序列分配基于碎片离子之间质量差的测量,因为除具有完全相同质量的两种氨基酸(分类为真正的同量异序):亮氨酸和异亮氨酸外,大部分氨基酸残基具有不同的质量。由于它们具有完全相同的质量和化学组成,因此它们在序列中的分配可以不太容易并通常报告为“x”或i/l。类似地,小氨基酸残基的一些特定组合可以与较大的氨基酸残基具有相同的质量。
5.就i/l残基而言,在序列中区分这两种同量异序氨基酸的能力可以在一些情况下是至关重要的。由于可以通过质谱确定蛋白序列,因此确定序列中正确同量异序氨基酸的性质的每种不确定性提高了“2k”倍的可能性数,其中“k”是蛋白序列中两种同量异序氨基酸之一的数目。如果蛋白测序的目标是提取信息以产生重组蛋白,则这些可能性数将直接
转化为基因数和因此要产生的基因引物。因此,分配正确的同量异序氨基酸的能力将减少要使用的引物数。这转化为显著的成本和时间的减少。
6.已发展了几种ms
‑
基方法来区分肽序列中的异亮氨酸(i)和亮氨酸(l)。早期方法依赖于使用更高的碰撞能,其中可以鉴别每种同量异序氨基酸的特异性的独特的碎片(falick等人1993)。然而,那些类型的ms仪在进行肽测序的实验室中很少使用或可用。其他方法包括不同的ms电离策略(nakamura等人1990;waern等人1978)或对于负模式检测的衍生化(ramsay等人1995;lindh等人2000),它们中的任一种在现代蛋白质组学实验室中都是不实用的。
7.对于肽碎片化,在蛋白质组学中使用的大部分ms仪以低碰撞能操作,因此可以不易于用于区分两种同量异序残基。近期,armirotti及其同事使用低能量型仪器做出了工作(armirotti等人,2007)。他们的方法基于特定低质量离子(亚胺离子)特征的检测。这种方法使得能够报告同量异序氨基酸之一的存在,然而,该方法不可以用于容易地精确定位正确氨基酸在序列中的位置,特别是在其中两种同量异序氨基酸存在于相同序列中的情况。
8.最近,lebedev和同事已发表了解决鉴别肽序列中正确的同量异序氨基酸所造成的困难的论文(zhokhov等人2017;lebedev等人2014)。他们基本上依赖于检测特定离子片段,w离子,这使得能够区分异亮氨酸和亮氨酸。他们依赖于产生c和z离子的电子传递解离(etd)的使用。后者可以在一定条件下进一步碎片化以产生对于亮氨酸和异亮氨酸具有不同的m/z特征的w离子。尽管该方法对于胰蛋白酶作为蛋白酶的使用是可接受的,并且可以在一些情况下在非胰蛋白酶肽中起作用以区分异亮氨酸和亮氨酸,但是大量异亮氨酸和亮氨酸将不易于分配给那些非胰蛋白酶肽。
9.尽管胰蛋白酶是大部分蛋白质组学工作所选的主要蛋白酶,但是蛋白的完整测序通常需要使用具有不同蛋白质切割规则的不同的蛋白酶以获得重叠的肽序列或者简单地以提高检测另外不易于可检测的序列部分的机会。当使用其他蛋白酶时,经常观察到的一个主要缺点在于由c末端碎片化所产生的离子质量较差。由于使用其他蛋白酶无法获得的所产生的离子质量等,因此胰蛋白酶是蛋白质组学市场中的主要蛋白酶。
10.已发展了几种肽衍生化策略来改变肽性质,包括提高它们的电离效率(mirzaei&regnier,2006)、它们的荷电状态(frey等人,2013;krusemark等人2011;perkins&fischer 2010)或者简单地允许它们定量(例如,可商购的方法,如icat、itraq和tmt)。大部分常规方法针对伯胺基团(即肽n末端或赖氨酸侧链)或者硫醇基团(半胱氨酸侧链)。然而,由于其低反应性,已很少研究肽的c末端衍生化。
11.在已对于c末端衍生化发展的少数方法中,最常见的方法是羧酸官能的酯化。然而,这种修饰导致了荷电状态的中和(goodlett等人,2001)。frey等人(2013)已发展了基于使用(7
‑
氮杂苯并三唑
‑1‑
基氧)三吡咯烷磷鎓六氟磷酸盐(pyaop)作为偶联剂的使用伯胺的c末端酰胺化的方法,从而在胰蛋白酶肽的电子传递解离(etd)下提高荷电状态和碎片化两者。另一种方法基于使用n
‑
(3
‑
二甲基氨丙基)
‑
n'
‑
碳酰二亚胺盐酸盐(edc)和1
‑
羟基
‑7‑
氮杂苯并三唑(hoat)来偶联2
‑
氨乙基三甲基氨(aetma)(ko&brodbelt,2012)。然而,偶联剂hoat在一定条件下显示出一定的爆炸性,因此需要谨慎处理。
12.肽c末端的化学偶联策略仅是少数综述的主题(例如,al
‑
warhi等人,2012)并且这些中很少用于蛋白质组学背景。已研究了使用不同的酶,特别强调使用羧肽酶y(cpy)的酶
促反式
‑
肽化(peptidation)。然而,就转肽作用而言,cpy显示出强序列特异性偏差,并且已使用简单的肽混合物(breddam等人,1980)或者蛋白(xu等人2011)进行了大部分研究。
13.照此,极少的策略可用于解释两种同量异序氨基酸:亮氨酸和异亮氨酸。因此,本发明人设法发展了当对肽测序时,用于区分同量异序氨基酸的方法。
技术实现要素:
14.本发明的目标是提供在多肽测序期间用于区分同量异序氨基酸的改善的方法。
15.本发明涉及特别是当使用胰蛋白酶以外的酶或切割策略时,在要通过质谱分析的肽的c末端侧添加碱性基团,arg
‑
c和lys
‑
c。这使得能够检测通常通过质谱仪不可检测的肽片段。这些碎片离子包括w离子,其然后可以用于解决由同量异序氨基酸,如i/l或者氨基酸组合,如分别对于n和q的gg和ga,所造成的可能的测序解释矛盾。
16.因此,本文提供了在多肽测序期间用于区分同量异序氨基酸和氨基酸组合:天门冬酰胺和甘氨酸
‑
甘氨酸;谷氨酰胺和甘氨酸
‑
丙氨酸;和/或谷氨酰胺和丙氨酸
‑
甘氨酸的方法,其包括以下步骤:
17.获得感兴趣的肽和/或用蛋白酶或化学切割法水解感兴趣的多肽以产生较短的肽;
18.在添加碱性官能团(正电荷)的条件下,将所获得和/或所产生的肽与能够使肽的游离c末端羧酸官能衍生化的偶联剂反应;
19.选择2+或以上的荷电状态,并且在质谱仪中在对于产生至少w离子有效的条件下使衍生化的肽碎片化;和
20.通过质谱检测w离子,并且鉴别掺入碱性官能团的额外质量的衍生化的肽;
21.其中,将正电荷添加至肽和/或多肽的游离c末端羧酸官能,并且分析w离子以区分同量异序氨基酸和以下氨基酸组合:异亮氨酸和亮氨酸;天门冬酰胺和甘氨酸
‑
甘氨酸;谷氨酰胺和甘氨酸
‑
丙氨酸;和/或谷氨酰胺和丙氨酸
‑
甘氨酸。
22.在所描述方法的某些实施方式中,反应步骤可以包括碱性官能团与至少多肽的c末端的化学或酶促偶联。
23.在其他实施方式中,偶联剂可以是碳二亚胺,例如,1
‑
乙基
‑3‑
(3
‑
二甲基氨丙基)碳二亚胺(edc)、二环己基碳二亚胺(dcc)或者n,n'
‑
二异丙基癸基碳二亚胺(dic),或者磷鎓离子,如(苯并三唑
‑1‑
基氧)三(二甲基氨)六氟磷酸磷鎓(bop)、(苯并三唑
‑1‑
基氧)三(吡咯烷并)六氟磷酸盐磷鎓(pybop)、(7
‑
氮杂苯并三唑
‑
基氧)三吡咯烷六氟磷酸磷鎓(pyaop),或者铵离子,如n
‑
[(1h
‑
苯并三唑
‑1‑
基)(二甲基氨)亚甲基]
‑
n
‑
甲基甲铵六氟磷酸盐n
‑
氧化物(n
‑
hbtu)、n
‑
[(二甲基氨)
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶
‑1‑
基亚甲基]
‑
n
‑
甲基甲铵六氟磷酸盐n
‑
氧化物(n
‑
hatu)或者1
‑
(1
‑
吡咯烷基
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶
‑1‑
基
‑
亚甲基)吡咯烷鎓六氟磷酸盐n
‑
氧化物(hapyu)。其他偶联剂也将对于本领域那些技术人员是显而易见的。
[0024]
在其他实施方式中,添加至多肽的碱性官能团可以是阳离子,如仲、叔或季胺或者胍基。例如,碱性官能团可以是3
‑
二甲基氨基
‑1‑
丙胺(3
‑
dmp)或精氨酸甲酯(metarg)、在c末端具有甲酯的二肽精氨酸
‑
精氨酸(metarg
‑
arg)、4
‑
(三甲胺)丁胺、5
‑
(二甲胺)戊胺、4
‑
(2
‑
氨乙基)吗啉、n,n
‑
二乙基
‑
1,4
‑
丁二胺、n,n
‑
二异丙基
‑
1,5
‑
戊二胺、4
‑
(3
‑
氨基丙基)吗
啉、n,n
‑
二甲基二丙烯三胺、3
‑
(二乙基氨基)丙胺、2
‑
氨基
‑5‑
二乙基氨基戊烷,或者磷鎓离子,如(3
‑
氨基丙基)(三苯基)溴化膦或者锍离子,如(3
‑
氨基
‑3‑
羧基丙基)二甲基锍。其他碱性官能团也将对于本领域那些技术人员是显而易见的。
[0025]
在其他实施方式中,添加剂可以在反应步骤中与偶联剂一起使用以有利于衍生化,如乙基氰基(肟基)乙酸酯(oxyma)、(1
‑
氰基
‑2‑
乙氧基
‑2‑
氧代亚乙基氨基氧基)二甲基氨基
‑
吗啉代
‑
碳鎓六氟磷酸盐(comu)、2
‑
氰基
‑2‑
(肟基)乙酸乙酯,钾盐(k
‑
oxyma)或者1
‑
[双(二甲基氨)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐(hatu)。其他添加剂也将对于本领域那些技术人员是显而易见的。
[0026]
在所描述方法中,可以实施其他步骤,其中(例如)在使赖氨酸基团转化为高精氨酸基团有效的条件下,使用o
‑
甲基异脲,通过胍基化来封闭多肽上的赖氨酸残基。赖氨酸侧链还可以二甲基化或二乙基化。在那些情况下,保留了赖氨酸的荷电状态。另一方面,可以通过乙酰化或丙酰化中和赖氨酸侧链的正电荷。最终,赖氨酸残基可以保持未修饰。通过在衍生化之前以这种方式修饰或封闭赖氨酸,有可能减少副反应,因为赖氨酸的伯胺基通常是使用一些上述偶联剂或添加剂的副反应的目标。它还降低了形成共价肽
‑
肽键(肽与肽而不是官能团与肽)的概率,尽管通常与肽相比,添加了明显摩尔过量的官能团以降低肽
‑
肽偶联的概率。
[0027]
在方法的其他实施方式中,可以包括在多肽的n末端封闭游离氨基的其他步骤。举例来说,可以用二碳酸二叔丁酯封闭多肽的n末端,或者简单地,使其作为赖氨酸残基二甲基化、二乙基化、乙酰化或丙酰化。如果包括,则该步骤通常将与赖氨酸修饰同时进行。
[0028]
还可以将净化步骤引入方法,借此净化衍生化的肽,例如,使用色谱柱或液相分离。例如,可以通过固相萃取(spe),使用反相、正相或离子交换spe或hplc对衍生化的肽进行净化。作为另外一种选择或者另外,可以通过液相分离对感兴趣的多肽和/或所衍生化的肽进行净化,例如,使用水饱和的乙酸乙酯,并干燥,例如,在低压下使用浓缩器,或者使用其他有机溶剂,如氯仿、二氯甲烷或乙醚。
[0029]
在所描述方法的实施方式中,赖氨酸(k)残基可以保持完整或(如果封闭)可以(例如)由于胍基化而具有42.021798da(1c 2n 2h)的额外质量,或者由于n末端和赖氨酸二甲基化(+28.0313da 2c 4h)、乙酰化(+42.0106da 2c 2h 1o)或丙酰化(+56.0262da 3c 4h 1o)具有额外质量。
[0030]
在所描述方法的其他实施方式中,多肽的天冬氨酸(d)和谷氨酸(e)残基酸以及c末端羧酸官能将(例如)由于metarg偶联而具有170.116761 da(4n 7c 14h 1o)的额外质量,或者使用3
‑
dmp而具有84.1051da(2n 5c 14h
‑
(2h 1o))的额外质量,或者使用5
‑
(二甲基氨)戊胺而具有112.136433da(2n 7c 18h
‑
(2h 1o))的额外质量,或者使用arg
‑
arg
‑
omet而具有326.2179da(13c 26h 8n 2o)的额外质量。
附图说明
[0031]
根据其中参考了附图的以下描述,本发明的这些及其他特征将变得更加显而易见,其中:
[0032]
图1显示了使用2种不同的蛋白酶,胰蛋白酶(左图)和胃蛋白酶(右图)所产生的肽的比较,以及对于两种蛋白酶,所选的肽碎片离子的ms/ms检测限。就胃蛋白酶水解来说,由
于无离子产生,更具体地,如果那些碎片不含碱性残基,如k、r或h(通过图右侧的红色柱所示),则一些c末端碎片是不可检测的。
[0033]
图2显示了本文所描述的方法的实施方式,包括当使用胃蛋白酶作为水解蛋白酶时,应用于图1中所示的相同蛋白的精氨酸甲酯(metarg)标记法(左图)。在右侧对于可检测的肽用绿色或者如果由于缺少电离而不可检测的用红色显示了理论产生的离子(从n末端或c末端肽末端)的“检测限”。
[0034]
图3显示了本文所描述的方法的实施方式,包括短假想肽“gkal”的metarg标记法的实例。蓝色化学结构显示了赖氨酸和肽n末端的二甲基化衍生化(减少潜在的副反应的任选的步骤)。红色的化学结构显示了添加至肽c末端的精氨酸甲酯基团(metarg)。metarg基团也将添加至大部分天冬氨酸和谷氨酸侧链。
[0035]
图4显示了肽fdklkhlvdepqnl(seq id no:1)的ethcd ms/ms光谱。肽仅在n末端和赖氨酸进行了二甲基化。肽在890.51amu为2+(1.68ppm)。对于c末端未标记形式,在ms/ms光谱中不存在所观察到的短c末端碎片离子(产生了极少的c末端碎片离子,仅检测到z11、z12和z13并且还检测到了少量c6至c13的c离子)。还作为3+和4+发现了相同肽的几种形式,其显示了类似的碎片化模式。在图的左上方,用绿色显示了从c末端侧产生的离子(z
‑
离子),而用紫色显示了从n末端侧产生的离子(c
‑
离子)。
[0036]
图5显示了与图4中相同,但在c末端用精氨酸甲酯另外标记的肽的ethcd ms/ms光谱。对整个序列观察到了从c末端侧更好的离子覆盖度。
[0037]
图6显示了图5的100至600amu尺度的放大图,以通过实验显示3种不同的同量异序的不确定物的分辨。具有43.055amu的质量变化的z1
‑
>w1离子使得能够鉴别肽c末端侧的亮氨酸(而不是异亮氨酸),而具有44.0141amu的质量变化的z2
‑
>w2使得能够分辨c末端倒数第二位的天门冬酰胺而不是gg的存在,并且最后,具有58.0309amu的质量变化的z3至w3使得能够确认在距c末端的第3位的谷氨酰胺,而不是2个残基丙氨酸和甘氨酸的存在。红色箭头显示了成对的z向w的转换。
[0038]
图7显示了从bsa的糜蛋白酶水解所产生的3种不同的肽,所有二甲基化均仅在n末端和赖氨酸残基侧链发生(左侧3个光谱)还在c末端用metarg标记了相同的肽(右侧3个光谱)。以hcd模式进行分析。另外,柱上实施标记(evosep尖头)并且直接加载到质谱仪上。比较显示出所产生的c末端离子的更好的覆盖度(通过每个序列中存在的红线显示)。
[0039]
图8显示了在c末端用metarg标记的肽feklgeygfqnal(seq id no:5)的ethcd ms/ms光谱。观察到来自c末端的优良离子覆盖度。从所产生的w离子鉴别了来自c末端位置1的亮氨酸l。
[0040]
图9显示了用3
‑
dmp标记的所标记的肽lqqcpfde(seq id no:6)的ethcd ms/ms光谱。由于c末端标记和d门冬氨酸残基的标记,观察到来自c末端的优良序列覆盖度。
[0041]
图10显示了就亮氨酸和异亮氨酸来说,在z离子的n末端产生的z和w离子的化学性质。尽管两种残基具有相同组成,但是可以通过它们的w离子区分它们。
[0042]
图11:(a)显示了以ethcd分析的假想肽序列gvi/last(模拟光谱,非真实实验)。在a中,未标记肽,可电离碎片仅来自n末端(模拟的c离子)。由于不存在可电离基团,因此对于该序列类型,不易产生z或w离子。在(b)中,序列为gvlast并且用精氨酸甲酯(metarg)在c末端标记,从而添加了170.116671amu的额外质量。n末端和c末端碎片两者(在该情况下,c和z
离子分别是可检测的)。额外的离子突出显示(w离子)为43.05amu的失去,从而确认在z4的n末端存在亮氨酸。在(c)中,显示了类似的肽,其中在z4离子碎片的n末端,亮氨酸被异亮氨酸替代。所产生的w离子作为替代显示了29.04amu的失去。除了由于假想的z4离子的n末端侧的i/l而不同的w离子碎片外,b和c两者的光谱是类似的。
[0043]
图12显示了对于以下的肽以ethcd模式采集的ms光谱:yggfl(seq id no:7);作为在26.9min洗脱的556.2769amu的1+离子(图12b),或者由于用精氨酸
‑
精氨酸甲酯在c末端标记(yggflrr
‑
omet),分别作为441.7513amu和294.8368amu且在14.8min洗脱的2+和3+离子(图12c)。441,7513amu峰的ethcd中的msms光谱显示了442.3047amu的z3离子,然后399.2480amu的强峰对应于43.0567amu的失去,它是亮氨酸的卫星w3离子(w3是lrr
‑
met)。
具体实施方式
[0044]
本发明人已开发了多肽测序方法,其改善了c末端碎片离子的产生并且另外使得能够区分同量异序氨基酸。方法包括肽标记步骤以提高c末端荷电状态和c末端碎片电离,具体地使得能够获得w
‑
离子并用于区分可能的同量异序氨基酸或难以分辨氨基酸组合的其他问题。
[0045]
如所描述的c末端标记与胃蛋白酶酶解水解的组合是特别有利的,因为它使得能够获得z1碎片(w1)。胃蛋白酶通常优先切割亮氨酸的c末端侧,这使得更易于确认该特定氨基酸。图2显示了方法如何改善碎片检测限的示意性概述。
[0046]
具体地,如下所示,方法包括在多肽测序期间区分同量异序氨基酸和氨基酸组合:天门冬酰胺和甘氨酸
‑
甘氨酸;谷氨酰胺和甘氨酸
‑
丙氨酸;和/或谷氨酰胺和丙氨酸
‑
甘氨酸:
[0047]
获得感兴趣的肽和/或用任何蛋白酶水解感兴趣的多肽,蛋白酶如(但不限于)胃蛋白酶、asp
‑
n、糜蛋白酶、lys
‑
c、弹性酶、glu
‑
c、嗜热菌蛋白酶或胰蛋白酶,或者使用化学切割产生感兴趣的多肽,如(但不限于)溴化氰(cnbr)、2
‑
(2
‑
硝基苯硫基)
‑
3h
‑
吲哚(bnps
‑
甲基吲哚)、甲酸、2
‑
硝基
‑5‑
氰硫基
‑
苯甲酸(ntcb)、1
‑
氰基
‑4‑
二甲基氨基吡啶鎓四氟硼酸盐(cdap)以从蛋白产生短肽;
[0048]
然后,使肽与能够使多肽的游离c末端羧酸官能衍生化的偶联剂在添加碱性官能团有效的条件下反应(其包括肽的c末端以及天冬氨酸和谷氨酸的侧链);
[0049]
选择2+或以上的荷电状态,并且在质谱仪中在对于产生至少w离子;优选地y、z和w离子有效的条件下使衍生化的肽碎片化;和然后,使用质谱检测w离子,优选地y、z和w离子,并且鉴别掺入碱性官能团的额外质量的衍生化的肽。
[0050]
将正电荷添加至多肽的游离c末端羧酸官能,并且分析w离子以区分同量异序氨基酸和氨基酸组合。具体地,方法使得能够区分:异亮氨酸和亮氨酸;天门冬酰胺和甘氨酸
‑
甘氨酸;谷氨酰胺和甘氨酸
‑
丙氨酸;和/或谷氨酰胺和丙氨酸
‑
甘氨酸。
[0051]
由于异亮氨酸和亮氨酸具有完全相同的质量(相同的化学组成),反映它们在肽序列中存在的常见的碎片离子,如b、c、y、z离子是相同的,因此那些离子将不能允许区分那2种氨基酸。然而,由于它们的不稳定性质,z和a离子产生二次离子(分别为w和d),其中(例如)z离子n末端的亮氨酸可以失去丙基(
‑
43.0548amu 3c 7h)并且异亮氨酸可以失去乙基(
‑
29.0391amu 2c 5h)或在较小程度上失去甲基(
‑
15.0235amu ch3)。因此,从亮氨酸或异
亮氨酸所产生的w离子具有不同的质量,因此允许在那2种氨基酸之间做出正确区分。以类似的方式,2个残基的甘氨酸
‑
甘氨酸(同位素质量为114.0429amu的gg)与单个残基天门冬酰胺(同位素质量为114.0429amu的n)具有相同的质量。由于它们较小的侧链,2个残基的gg在z离子的n末端不产生w离子,而另一方面,天门冬酰胺(n)在z离子的n末端将失去44.0136amu以产生w离子(失去1c 1o 1n 2h)。以类似的方式,谷氨酰胺残基(同位素质量为128.0586amu的q)与2个残基的甘氨酸丙氨酸的组合(同位素质量为128.0586amu)具有相同质量。2个残基的甘氨酸和丙氨酸的侧链也过小,从而不会产生任何w离子,然而谷氨酰胺在z
‑
离子的n末端将失去58.0293amu以产生w离子(失去s 2c 1n 1o 4h)。在所描述实例中,如果位于z
‑
离子的n末端,则较大的残基(i、l、n或q中的任一种)将失去它们大部分的侧链,从而留下较短的序列,就亮氨酸、天门冬酰胺和谷氨酰胺来说,通常双键ch2。如果残基一开始在它们的侧链中具有至少2个碳(甘氨酸和丙氨酸不是这种情况),则那些失去仅是可能的。最终,为了具有w
‑
离子,需要具有z
‑
离子,并且为了具有z
‑
离子,则必须在c末端存在正电荷。这使用(例如)通过metarg的衍生化完成。
[0052]
图10显示了从亮氨酸和异亮氨酸所产生的w离子的实例。图11,显示无标记的短肽(图11a)和在从肽n末端数的第3位具有使用亮氨酸的c末端标记的相同肽(图11b)或者这种情况下,在从肽n末端数的第3位具有使用异亮氨酸的c末端标记的相同肽的ms/ms光谱的原理图。图11b和c中的2种光谱是非常类似的,仅有与序列中亮氨酸或异亮氨酸的存在有关的微小显示差异。在以下所讨论的实验中更详细地讨论了这种情况。
[0053]
在方法的具体实施方式中,如所描述的肽的衍生化有利于肽序列中异亮氨酸vs.亮氨酸的正确分配,特别是在其中将胰蛋白酶以外的蛋白酶用于水解的情况下。方法的实施方式还使得能够区分由具有相同质量的氨基酸残基和组合所造成的其他不确定性。
[0054]
因此,所描述方法有利于:
[0055]
i.通过在c末端不具有碱性残基的肽质谱的c末端测序,和
[0056]
ii.区分同量异序氨基酸,如异亮氨酸和亮氨酸,或者同量异序氨基酸组合,如与2个甘氨酸(gg)具有相同质量的天门冬酰胺(n),和与丙氨酸和甘氨酸(ag或ga)具有相同质量的谷氨酰胺(q)。由于w离子的产生,这种区分是可能的,w离子是由于在肽的c末端添加带正电的官能团所产生的。
[0057]
如以下进一步详细描述的,可以通过w
‑
离子的检测,例如,使用ethcd(电子转移/高能碰撞解离)、热电子捕获解离(hecd)、etd
‑
hcd
‑
ms3、活化离子电子转移解离(ai
‑
etd)或者ai
‑
etd结合使用红外多光子激活的后激活(post
‑
activation using infrared multiphoton activation)(ai
‑
etd+)或者补充了uv
‑
光解离的etd(etuvpd)或者简单地uv
‑
光解离(uvpd)来解释所有这些同量异序组合。
[0058]
所描述方法包括对从肽c末端碎片所产生的离子的分析。可以使用特异性蛋白酶—胰蛋白酶
‑
其通过在精氨酸和赖氨酸的c末端侧的切割将蛋白切割成短肽来产生这些离子。精氨酸和赖氨酸是碱性残基,其在酸性条件下带有正电荷,这是通过质谱的正离子检测的基本性质。通过其他蛋白酶相对很少产生c末端离子,因此使用除胰蛋白酶以外的蛋白酶来区分异亮氨酸和亮氨酸是更繁琐的并且依赖于接近于切割位点的碱性残基的存在,这对于大部分蛋白并不总是这种情况。图1显示了使用胰蛋白酶或胃蛋白酶对可以检测的离子和碎片的影响的图示。所提议的衍生化使得能够使用任何蛋白酶产生c末端离子,蛋白酶
如(但不限于)asp
‑
n、glu
‑
c、糜蛋白酶、弹性酶、胰蛋白酶
‑
n、lys
‑
n和胃蛋白酶。
[0059]
基于产生w
‑
离子的如本文所描述的质谱法的使用结合标记或肽衍生化步骤,使得能够在任何肽的c末端添加正电荷而不依赖于所使用的蛋白酶或化学切割。这使得方法能够区分异亮氨酸和亮氨酸及其他同量异序氨基酸组合,这在多肽测序期间是特别困难的。
[0060]
定义:
[0061]
术语“官能团”是指可以在肽的c末端与羧基偶联并且具有游离碱性基团的任何分子。这可以无限制地包括阳离子,如仲、叔或季胺,或者胍基(例如,3
‑
二甲基氨基
‑1‑
丙胺(3
‑
dmp)或精氨酸甲酯(metarg)、4
‑
(三甲胺)丁胺、5
‑
(二甲胺)戊胺、4
‑
(2
‑
氨乙基)吗啉、n,n
‑
二乙基
‑
1,4
‑
丁二胺、n,n
‑
二异丙基
‑
1,5
‑
戊二胺、4
‑
(3
‑
氨基丙基)吗啉、n,n
‑
二甲基二丙烯三胺、3
‑
(二乙基氨基)丙胺、2
‑
氨基
‑5‑
二乙基氨基戊烷,磷鎓离子,如(3
‑
氨基丙基)(三苯基)溴化膦或者锍离子,如(3
‑
氨基
‑3‑
羧基丙基)二甲基锍。
[0062]
术语“偶联剂”是指能够使感兴趣的肽的c末端羧酸官能,以及任选地肽序列中的任何谷氨酸和门冬氨酸残基的羧酸官能衍生化,借此增加上述官能团的试剂。偶联剂的实例包括(但不限于)碳二亚胺(例如,1
‑
乙基
‑3‑
(3
‑
二甲基氨丙基)碳二亚胺(edc))、二环己基碳二亚胺(dcc)或者n,n'
‑
二异丙基癸基碳二亚胺(dic),或者磷鎓离子,如(但不限于)(苯并三唑
‑1‑
基氧)三(二甲基氨)六氟磷酸磷鎓(bop)、(苯并三唑
‑1‑
基氧)三(吡咯烷并)六氟磷酸盐磷鎓(pybop)、(7
‑
氮杂苯并三唑
‑
基氧)三吡咯烷六氟磷酸磷鎓(pyaop),或者铵离子,如n
‑
[(1h
‑
苯并三唑
‑1‑
基)(二甲基氨)亚甲基]
‑
n
‑
甲基甲铵六氟磷酸盐n
‑
氧化物(n
‑
hbtu)、n
‑
[(二甲基氨)
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶
‑1‑
基亚甲基]
‑
n
‑
甲基甲铵六氟磷酸盐n
‑
氧化物(n
‑
hatu)、1
‑
(1
‑
吡咯烷基
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶
‑1‑
基
‑
亚甲基)吡咯烷鎓六氟磷酸盐n
‑
氧化物(hapyu)。
[0063]
术语“添加剂”是指通常与偶联剂一起在衍生化反应中使用以提高反应性的试剂。添加剂的实例包括(但不限于)乙基氰基(肟基)乙酸酯(oxyma)、(1
‑
氰基
‑2‑
乙氧基
‑2‑
氧代亚乙基氨基氧基)二甲基氨基
‑
吗啉代
‑
碳鎓六氟磷酸盐(comu)、2
‑
氰基
‑2‑
(肟基)乙酸乙酯,钾盐(k
‑
oxyma)和1
‑
[双(二甲基氨)亚甲基]
‑
1h
‑
1,2,3
‑
三唑并[4,5
‑
b]吡啶鎓3
‑
氧化物六氟磷酸盐(hatu)。
[0064]
实施例:
[0065]
(i)官能团分析:
[0066]
如以上所讨论的,可以在所描述方法中使用的官能团的类型可以是不同的,只要它们可以偶联至肽的游离羧基并且具有游离碱性基团。测试了3种不同的官能团(分子):3
‑
二甲基氨基
‑1‑
丙胺(3
‑
dmp)、精氨酸甲酯(metarg)和二肽精氨酸
‑
精氨酸
‑
甲酯(rr
‑
omet)。
[0067]
在本实施例中,使用偶联剂1
‑
乙基
‑3‑
(3
‑
二甲基氨丙基)碳二亚胺(edc),以乙基氰基(肟基)乙酸酯(oxyma)作为添加剂/强化剂,实施官能团和感兴趣的肽之间的偶联。
[0068]
使用这些试剂,将使任何游离羧酸官能衍生化,并因此肽序列中的肽c末端氨基酸以及任何谷氨酸和门冬氨酸将与下列化合物偶联:(a)如果使用metarg,精氨酸甲酯—质量提高170.116761da(4n 7c 14h 1o),或者(b)如果使用3
‑
dmp,3
‑
二甲基氨基
‑1‑
丙胺,质量提高84.105133da(2n 5c 12h
‑
1o),或者(c)使用rr
‑
omet,精氨酸
‑
精氨酸
‑
甲酯,质量提高326.2179da(13c 26h 8n 2o)。就3
‑
dmp和metarg来说,将每个偶联的官能团1单位的正电荷升高添加至标记的肽,而就rr
‑
omet来说,将每个偶联的官能团2单位的升高添加至标记的
肽。
[0069]
(ii)标记程序:实施例1
[0070]
将以下所提供的整体程序应用于从胃蛋白酶水解所产生的10ug肽,然而,它可以应用于更小或更大量的肽混合物并因此缩放。它还可以应用于由任何其他蛋白酶或化学切割的使用所产生的任何其他的肽混合物。
[0071]
1.用胍基化基团封闭赖氨酸(任选的):在本发明的情况下,本发明人使用了edc/oxyma,其可以产生几个副反应。最多的副反应之一是赖氨酸胍基化(将赖氨酸修饰为高精氨酸)。这种具体的修饰不影响赖氨酸的电离状态,然而,这种副反应通常是部分的,因此增加了另外水平的不必要的复杂性。因此,在所描述方法中设想了任选的步骤以使用o
‑
甲基异脲,在ph 10.5用胍基封闭所有赖氨酸残基,因此所有赖氨酸然后被高精氨酸替代。向10ug肽水解物中添加50ul 0.1g/ml o
‑
甲基异脲的溶液,并在65℃保持1h。然后,通过在反相固相萃取装置(rp
‑
spe)上净化肽混合物来终止反应以除去可以干扰后续标记步骤的额外的偶联剂。
[0072]
2.用二碳酸二叔丁酯,boc封闭游离n末端(任选的):向10ug干燥且净化的水解的肽中添加8ul boc和25ul水;在室温(rt)下超声并培育1h。如果过量添加且同时添加偶联剂和官能团,则该步骤不是必需的。
[0073]
3.净化:在spe(反相、正相或离子交换)上或通过使用液相分离净化肽。在这种情况下,使用水饱和的乙酸乙酯。简要地,向样品加入100ul mqw水,加入1ml水饱和的乙酸乙酯,良好混合,除去上层并用乙酸乙酯重复净化程序2
‑
3次以除去额外的boc试剂。然后,在低压下使用浓缩器干燥肽。
[0074]
4.肽偶联:将肽在100ul mqw h2o中复原,加入80ul精氨酸甲酯(32.5mg/ml在h2o和二甲基甲酰胺(dmf)的1:1的混合物中)。使用1n氢氧化钠将ph调节至5
‑
6,加入40ul edc(100mg/ml在dmf:水(950:50)中)以及26ul oxyma(100mg/ml在dmf中),再次使用1n naoh将ph调节至5
‑
6。使反应在rt培育过夜。然后,通过添加50ul三氟乙酸(tfa)并在rt培育1h来终止反应。
[0075]
5.肽净化:如之前在步骤3)中所描述,使用spe
‑
rp或水饱和的乙酸乙酯进行。
[0076]
6.通过lc
‑
ms的肽分析:大部分离子应具有大于1的荷电状态,因此可以通过甚至对于非胰蛋白酶解的肽选择2和以上的荷电状态来进行ms/ms的肽选择。搜索引擎的参数如下所示:由于胍基化,赖氨酸k可以具有42.021798amu的额外质量(1c 2n 2h)。由于精氨酸甲酯偶联,残基天冬氨酸(d)和谷氨酸(e)以及肽c末端可以具有170.116761da(4n 7c 14h 1o)的额外质量。将正电荷加入至任何所添加的标记的天冬氨酸、谷氨酸残基和c末端。另外,就使用精氨酸甲酯的任何肽标记来说,在189.134626amu(7c 4n 2o 16h 1h+)发现了ms/ms光谱中的报告离子,这使得能够确认该标记法是成功的。在图2中显示了整体标记程序,尽管图3显示了赖氨酸的化学结构向高精氨酸的转化以及在假想肽的c末端所添加的metarg。
[0077]
标记程序:实施例2
[0078]
将以下所提供的整体程序应用于从胃蛋白酶水解所产生的10ug肽。
[0079]
1.用二甲基基团封闭赖氨酸和肽n末端(任选的):在本发明的情况下,本发明人使用了edc/oxyma,其可以产生几个副反应。因此,在所描述方法中设想了另一个任选步骤以
使用甲醛封闭所有赖氨酸残基和肽n末端。将10ug肽水解物在5ul水:甲醇(1:1),1ul 37wt%的甲醛水溶液,3ul 8m硼烷吡啶复合物和3ul 4
‑
甲基吗啉中复原,培育1h,然后低压蒸发;可以通过以类似方式第二次标记来重复二甲基化步骤。然后,用甲酸使样品酸化并复原至100ul水中,用水饱和的乙酸乙酯清洗3次,然后低压干燥。如实施例1中所描述实施精氨酸甲酯偶联。
[0080]
2.肽偶联:将肽在100ul mqw h2o中复原,加入80ul精氨酸甲酯(32.5mg/ml在h2o和二甲基甲酰胺(dmf)的1:1的混合物中)。使用1n氢氧化钠将ph调节至5
‑
6,加入40ul edc(100mg/ml在dmf:水(950:50)中)以及26ul oxyma(100mg/ml在dmf中),再次使用1n naoh将ph调节至5
‑
6。使反应在rt培育过夜。然后,通过添加50ul三氟乙酸(tfa)并在rt培育1h来终止反应。
[0081]
3.肽净化:如之前在实施例1的步骤3)中所描述的,使用spe
‑
rp或水饱和的乙酸乙酯进行。
[0082]
4.通过lc
‑
ms的肽分析:大部分离子应具有大于1的荷电状态,因此可以通过甚至对于非胰蛋白酶解的肽选择2和以上的荷电状态来进行ms/ms的肽选择。搜索引擎的参数如下所示:由于二甲基化,赖氨酸k和肽n末端具有28.0313amu的额外质量(2c 4h)。由于精氨酸甲酯偶联,残基天冬氨酸(d)和谷氨酸(e)以及肽c末端可以具有170.116761da(4n 7c 14h 1o)的额外质量。将正电荷加入至任何所添加的标记的天冬氨酸、谷氨酸残基和c末端。另外,就使用精氨酸甲酯的任何肽标记来说,在189.134626amu(7c 4n 2o 16h 1h+)发现了ms/ms光谱中的报告离子,这使得能够确认该标记法是成功的。在图2中显示了整体标记程序,尽管图3显示了赖氨酸的化学结构向高精氨酸的转化以及在假想肽的c末端所添加的metarg。
[0083]
标记程序:实施例3:
[0084]
使用pyaop的c末端标记程序
[0085]
将以下所提供的整体程序应用于从胃蛋白酶水解所产生的10ug肽。
[0086]
1.封闭赖氨酸和肽n末端:这未进行,因为与来自肽的胺相比,摩尔过量加入胺标记试剂,因此显著降低了发生肽
‑
肽偶联的机会。
[0087]
2.c末端偶联反应:如下所示制备了胺溶液:将100mg甲酯精氨酸(metarg)溶于50ul水和26ul 4
‑
甲基吗啉(nmm)中。如下制备了偶联溶液:将66mg 7
‑
氮杂苯并三唑
‑1‑
基氧)三吡咯烷磷鎓六氟磷酸盐(pyaop)溶于145ul二甲亚砜(dmso)。向干燥的10ug肽中添加10ul dmso,然后添加14ul胺溶液和6ul偶联溶液。将反应在室温下过夜进行。
[0088]
3.反应淬灭和样品净化:将50ul tfa和100ul水加入至样品中,并保持1h。然后,使用3
×
1ml的水饱和的乙酸乙酯并使用1ml氯仿的另外一次清洗来净化样品以除去dmso。然后,使用低压离心机(speedvac)干燥样品,然后将干燥的标记的肽水解物在0.1%的甲酸水缓冲液中复原以用于lc
‑
ms分析。
[0089]
4.通过lc
‑
ms的肽分析:大部分离子应具有大于1的荷电状态,因此可以通过甚至对于非胰蛋白酶解的肽选择2和以上的荷电状态来进行ms/ms的肽选择。搜索引擎的参数如下所示:由于精氨酸甲酯偶联,残基天冬氨酸(d)和谷氨酸(e)以及肽c末端可以具有170.116761da(4n 7c 14h 1o)的额外质量。将正电荷加入至任何所添加的标记的天冬氨酸、谷氨酸残基和c末端。另外,就在c末端使用精氨酸甲酯的任何肽标记来说,在
189.134626amu(7c 4n 2o 16h 1h+)发现了ms/ms光谱中的报告离子,这使得能够确认该标记法是成功的。
[0090]
在固相萃取反相柱(spe)上使用edc
‑
oxymac的c末端标记程序:实施例4
[0091]
本实施例是牛血清白蛋白(bsa)10ug的糜蛋白酶水解物。将25mg固相萃取(spe)柱bond elut c18 lms 1cc 25mg珠体积(部件号no 12105021,来自agilent)打开并将反相装填介质在1ml乙腈中复原以产生浆液。将rp c18 lms烧结块(frits)切成小块并将一个小块引入200ul移液器尖头以起到烧结块/过滤器的作用以保留介质。使用100ul浆液并将其加入到200ul移液器尖头中的烧结块的上方。另一种策略包括直接使用evosep尖头而无任何改变,尽管对于自制的bond elut spe尖头,其结合能力接近于2ug肽而不是10ug。对尖头施加正压以迫使溶剂流动,而反相(rp)装填介质保持在烧结块上方。开始,将100ul甲醇加入至尖头并通过重力流动使其流过。加入50ul乙腈并用80ul水调节柱。将2
‑
10ug bsa肽水解物在50ul水中的溶液加入至调节好的spe尖头。使肽水解物保留在spe尖头上(自制的bond elut或者evosep尖头)并用40ul水清洗。如下所示制备了二甲基化溶液:将21ul 37%的甲醛和7.5mg氰基硼氢化物合并并将溶液加入1ml水。使150ul二甲基化溶液通过尖头,并将rp树脂在二甲基化溶液中在室温下浸泡过夜。
[0092]
用50ul h2o清洗spe尖头上的结合样品2次;制备了3种偶联溶液。溶液1:50mg edc在950ul dmf+50ul h2o中,溶液2:oxyma 100mg在100ul dmf中,溶液3:52mg metarg在400ul h2o+400ul dmf中。
[0093]
对于样品,使用了2种不同的柱上c末端标记方法:
[0094]
方法1:200ul h2o+26ul溶液1+40ul溶液2和80ul溶液3+20ul naoh 1n;将所有溶液混合在一起并使其流过柱。
[0095]
方法2:是2
‑
步法,其中步骤1是使用200ul h2o+26ul溶液1+40ul溶液2并在柱上保留5
‑
10min的羧酸活化;然后,步骤2是如下所示进行的偶联反应:将50ul(50ul溶液3+150ul h2o+10ul naoh)的混合物加入至spe尖头并将rp介质在该溶液中浸泡2h。可以再次重复活化和偶联步骤。
[0096]
然后,用3
×
50ul 0.1%的甲酸水溶液清洗样品;将rp尖头在eppendorf管中与0.1%fa水培育1h(以水解过量的偶联剂)。然后,用100ul乙腈洗脱标记的肽并低压干燥(speedvac),然后将干燥的标记的肽水解物在0.1%的甲酸水缓冲液中复原以用于lc
‑
ms分析。对于evosep尖头,在质谱仪上使用厂家建议的规程在线进行洗脱。
[0097]
通过lc
‑
ms的肽分析:大部分离子应具有大于1的荷电状态,因此可以通过甚至对于非胰蛋白酶解的肽选择2和以上的荷电状态来进行ms/ms的肽选择。搜索引擎的参数如下所示:由于精氨酸甲酯偶联,残基天冬氨酸(d)和谷氨酸(e)以及肽c末端可以具有170.116761da(4n 7c 14h 1o)的额外质量。将正电荷加入至任何所添加的标记的天冬氨酸、谷氨酸残基和c末端。另外,就使用精氨酸甲酯的任何肽标记来说,在189.134626amu(7c 4n 2o 16h 1h+)发现了ms/ms光谱中的报告离子,这使得能够确认该标记法是成功的。最终,由于二甲基化,赖氨酸k和肽n末端具有28.0313amu的额外质量(2c 4h)。柱上标记策略允许整体c末端标记法自动进行并且减少了溶剂,如乙酸乙酯的使用。
[0098]
标记程序:实施例4:使用pyaop和二肽试剂精氨酸
‑
精氨酸
‑
甲酯试剂的c末端标记程序:实施例4
[0099]
将以下所提供的整体程序应用于10ug合成肽亮氨酸脑啡肽(sigma aldrich l9133)。
[0100]
1.封闭赖氨酸和肽n末端:这未进行,因为与来自肽的胺相比,摩尔过量加入胺标记试剂,因此显著降低了发生肽
‑
肽偶联的机会。
[0101]
2.c末端偶联反应:如下所示制备了胺溶液:将100mg甲酯精氨酸
‑
精氨酸(arg
‑
arg
‑
omet)溶于50ul水和26ul 4
‑
甲基吗啉(nmm)中。如下制备了偶联溶液:将66mg 7
‑
氮杂苯并三唑
‑1‑
基氧)三吡咯烷磷鎓六氟磷酸盐(pyaop)溶于132ul二甲亚砜(dmso)。向干燥的10ug肽中添加10ul dmso,然后添加14ul胺溶液和6ul偶联溶液。将反应在室温下过夜进行。
[0102]
3.反应淬灭和样品净化:将320ul 0.1%tfa加入至样品,然后使用640ul氯仿,净化样品2次以除去dmso。然后,使用低压离心机(speedvac)干燥样品,然后将干燥的标记的肽水解物在0.1%的甲酸水缓冲液中复原以用于lc
‑
ms分析。
[0103]
4.通过lc
‑
ms的肽分析:大部分离子应具有大于1的荷电状态,因此可以通过甚至对于非胰蛋白酶解的肽选择2和以上的荷电状态来进行ms/ms的肽选择。搜索引擎的参数如下所示:由于精氨酸
‑
精氨酸甲酯偶联,残基天冬氨酸(d)和谷氨酸(e)以及肽c末端可以具有326.2179da(13c 26h 8n 2o)的额外质量。将至少2个正电荷加入至c末端。另外,就在c末端使用精氨酸
‑
精氨酸甲酯的任何肽标记来说,在189.134626amu(7c 4n 2o 16h 1h+)发现了ms/ms光谱中的报告离子,这使得能够确认该标记法是成功的。
[0104]
(iii)未标记和标记的肽的ms/ms分析:
[0105]
本发明人实施了相同的肽的ms/ms分析(在c末端未标记和在c末端用精氨酸甲酯标记,分别为图4和5)。在orbitral fusion仪上以ethcd模式(thermo
‑
fisher)采集数据。图4、5和6中所示的数据显示了标记对肽c末端碎片电离质量的影响。肽序列是来自牛血清白蛋白的由使用胃蛋白酶的蛋白酶水解所产生的肽:fdklkhlvdepqnl(seq id no:1)。在图4中,仅在n末端和赖氨酸对肽进行了二甲基化。肽在890.51amu为2+。
[0106]
对于图4中所示的c末端未标记形式,在ms/ms光谱中不存在所观察到的短c末端碎片,仅检测到z11、z12和z13并且还检测到了少量(c6至c13)的c离子。还作为3+和4+发现了相同肽的几种形式,其显示了类似的碎片化模式(数据未显示)。仍在该图4中,尽管胃蛋白酶在亮氨酸的c末端侧切割,但是z1或w1离子不可检测,从而毫无疑问地鉴定该亮氨酸位于肽的c末端。图5显示了用metarg标记后的相同的肽。所选光谱为在650.71amu,具有用精氨酸甲酯标记的c末端的肽的3+(另外,如图4中一样,用二甲基化在n末端和赖氨酸标记了肽)。从z1至z3、z5、z6、z8
‑
z13的更完整的c末端碎片化模式和更广泛的碎片系列是可检测的,这使得能够更好地确认序列内容物。图6是图5中100至600amu之间的范围的放大,其突出显示了特定离子z1、z2和z3以及它们分别的相关w1、w2和w3离子。这种短序列允许分辨3种不同的同量异序情况。243.1438amu的离子w1是来自z1离子的43.0555amu,因此确认亮氨酸在该肽c末端的存在(而不是异亮氨酸)。以类似方式,z2在44.0141amu的w2确认n(而不是gg)的存在,并且最终来自z3的58.0309amu的较低质量处的w3确认q而不是ag/ga序列的存在。不使用c末端标记的情况下,分辨那些同量异序情况将是不可能的。
[0107]
图7显示了对于以下3种肽以hcd模式采集的ms/ms光谱:
[0108]
avegpklvvstqtala(seq id no:2);在肽n末端和赖氨酸上二甲基化(左上图)以及用精氨酸甲酯(metarg)在c末端标记相同的肽(右上图)。
[0109]
dehvklvneltef(seq id no:3);在肽n末端和赖氨酸上二甲基化(左中图)以及用metarg在c末端标记相同的肽(右中图)。
[0110]
qeakdaflgsfly(seq id no:4);在肽n末端和赖氨酸上二甲基化(左下图)以及用metarg在c末端标记相同的肽(右下图)。
[0111]
肽来自bsa的糜蛋白酶水解,然后根据实施例4中详细说明的程序柱上标记(evosep)。所有3种肽显示使用metarg的c末端标记对c末端y
‑
离子的覆盖度更好(右侧3个光谱,通过序列中所示的红线显示,其与所检测的碎片相关)。
[0112]
图8显示了对于以下的肽以ethcd模式采集的ms/ms光谱:
[0113]
feklgeygfqnal(seq id no:5);作为581.31amu处的3+离子。也是在伯胺转化为二甲基化并且用精氨酸甲酯封闭c末端后以ethcd模式采集的序列id 5的光谱显示出c末端离子的优良覆盖度。分别与z1和w1有关的286.2015amu的碎片离子以及243.1434amu的碎片离子的存在确认了z1位置中亮氨酸的存在。肽的未标记形式不具有短z
‑
离子碎片(数据未显示)。最后一个实验光谱(图9)是作为标记试剂的3
‑
dmp对肽lqqcpfde(seq id no:6)的c末端侧的使用的实例。
[0114]
首先用二甲基化标记肽所有的伯胺基,并且在c末端和在残基d用试剂3
‑
dmp标记。另外,在胃蛋白酶水解之前,用碘乙酰胺使半胱氨酸烷基化。离子在411.57amu为3+。尽管与使用精氨酸甲酯所观察到的碎片化模式相比更差,但是用3
‑
dmp标记产生了少量在类似的肽的未标记形式中观察不到的c末端碎片。还突出显示作为c末端标记试剂的精氨酸似乎更有效地产生了c末端碎片,如y、z离子。
[0115]
图12显示了对于以下的肽以ethcd模式采集的ms光谱:yggfl(seq id no:7);作为在26.9min洗脱的556.2769amu的1+离子(图12b),或者由于用精氨酸
‑
精氨酸甲酯在c末端标记(yggflrrmet),分别作为441.7513amu和294.8368amu且在14.8min洗脱的2+和3+离子(图12c)。441,7513amu峰的ethcd中的msms光谱显示了442.3047amu的z3离子,然后399.2480amu的强峰对应于43.0567amu的失去,它是亮氨酸的卫星w3离子(w3是lrr
‑
met)。添加rr
‑
omet将肽的保留时间从26.9min显著降低至14.8min,这可以是检测高疏水性肽的有用策略。
[0116]
已通过举例说明描述了一种或多种目前优选的实施方式。对于本领域技术人员将显而易见的是可以在不背离如权利要求中所定义的本发明的范围的情况下做出一些改变和变化。
[0117]
参考文献:
[0118]
al
‑
warhi ti,al
‑
hazimi hma,el
‑
faham a(2012)recent development in peptide coupling reagents.journal of saudi chemical society 16,97
–
116.doi:10.1016/j.jscs.2010.12.006.
[0119]
armirotti a,millo e,damonte g.(2007)how to discriminate between leucine and isoleucine by low energy esi
‑
trap msn,j.am.soc.mass spectrom.,18,57
–
63.
[0120]
breddam k,widmer f,johansen jt(1980)carboxypeptidase y catalyzed transpeptidations and enzymatic peptide synthesis,carlsberg res.commun 45,237
‑
247.
proteins.methods mol biol.;753:77
‑
91.doi:10.1007/978
‑1‑
61779
‑
148
‑
2_6.
[0131]
lebedev at,damoc e,makarov aa,samgina ty(2014)discrimination of leucine and isoleucine in peptides sequencing with orbitrap fusion mass spectrometer.anal.chem.2014,86,7017
‑
7022 dx.doi.org/10.1021/ac501200h.
[0132]
lindh i,hjelmqvist l,bergman t,j,griffiths wj.(2000)de novo sequencing of proteolytic peptides by a combination of c
‑
terminal derivatization and nano
‑
electrospray/collision
‑
induced dissociation mass spectrometry.journal of the american society for mass spectrometry 11(8),673
‑
686 https://doi.org/10.1016/s1044
‑
0305(00)00138
‑
0.
[0133]
oh hb,moon b.(2015)radical
‑
driven peptide backbone dissociation tandem mass spectrometry.mass spectrom rev.;34(2):116
‑
32.doi:10.1002/mas.21426.
[0134]
ma b.(2015)novor:real
‑
time peptide de novo sequencing software.j am.soc of mass spectrom.26(11)1885
‑
1894.
[0135]
ma m,kutz
‑
naber kk,li l.(2007)methyl esterification assisted maldi ftms characterization of the orcokinin neuropeptide family.anal.chem,79 673
‑
681.
[0136]
medzihradszky kf&chalkley rj.(2016)lesson i de novo peptide sequencing by tandem mass spectrometry,mass spectrom.rev.;34(1):43
–
63.
[0137]
mirzaei h and regnier f.(2006)enhancing electrospray ionization efficiency of peptides by derivatization.anal.chem.78,12,4175
‑
4183 doi:10.1021/ac0602266.
[0138]
nakamura t,nagaki h,ohki y,kinoshita t.(1990)differentiation of leucine and isoleucine residues in peptides by consecutive reaction mass spectrometry.anal.chem.,62(3),311
–
313.
[0139]
perkins,p.d.,fischer,s.m.(2010)peptide derivatization method to increase fragmentation information from ms/ms spectra.patent publication no us 2010/7,838,303 b2
[0140]
ramsay sl,steinborner st,waugh rj,dua s,bowie jha.(1995)simple method for differentiating leu and ile in peptides.the negative
‑
ion mass spectra of[m h]ions of phenylthiohydantoin leu and ile.rapid commun.mass spectrom.,9(13),1241
–
1243.
[0141]
riley nm&coon jj(2018)the role of electron transfer dissociation in modern proteomics.anal.chem.90,40
‑
64.doi:10.1021/acs.analchem.7b04810
[0142]
wysocki vh,resing ka,zhang q,cheng g.(2005)mass spectrometry of peptides and proteins.methods.;35(3):211
‑
22.epub 2005 jan 20.review.
[0143]
xiao y,vecchi mm,wen d.(2016)distinguishing between leucine and isoleucine by integrated lc
‑
ms analysis using an orbitrap fusion mass spectrometer.anal chem.2016 nov 1;88(21):10757
‑
10766.epub 2016 oct 14.
[0144]
xu g,shin sby,jaffrey sr.(2011)acs chem biol;6(10),1015
‑
1020.doi:10.1021/cb200164h.
[0145]
zhokhov ss,kovalyov sv,samgina ty,lebedev at.(2017)an ethcd
‑
based method for discrimination of leucine and isoleucine residues in tryptic peptides.j am soc mass spectrom;28(8):1600
‑
1611.doi:10.1007/s13361
‑
017
‑
1674
‑
3.epub 2017 apr 26.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1