一种外周血染色体G带的制作方法与流程
一种外周血染色体g带的制作方法
技术领域
1.本发明涉及医学检验技术领域,具体涉及一种外周血染色体g带的制作方法。
背景技术:
2.染色体是基因的载体,染色体异常,势必引起基因表达不平衡,从而引起一系列的临 床症状。研究表明染色体异常与血液系统疾病、智力低下、生殖异常等密切相关。在人类生 殖研究领域,流产、死胎、胎停不孕不育、无精少精、卵巢早衰、幼稚子宫、出生缺陷、第 二性征发育不全等疾病与染色体的结构异常、数目异常以及多态性相关,因此对此类患者进 行染色体核型分析很有必要。1968年瑞典细胞化学家caspersson等应用荧光染料氮芥喹吖因 (quinacrine mustard,qm)处理染色体后,在荧光显微镜下可观察到染色体沿其长轴显示出 一条条宽窄和亮度不同的横纹,即染色体的显带(band)。随后又出现了其他几种染色体显带 技术。显带技术可将人类的染色体显示出各自特异的带纹,称为带型。g带是目前被广泛应用 的一种带型,它主要是被姬姆萨(giemsa)染料染色后而显带,故称之为g显带技术,其所显示 的带纹分布在整个中期细胞的染色体上。g显带方法简单,带纹清晰,染色体标本可长期保存, 因此被广泛用于染色体病的诊断和研究。
[0003][0004]
外周血染色体的制作主要包括:培养、收获、低渗、固定、分带、染色及制片等步骤, 每个步骤都可能会影响外周血染色体的制备成功。由于外周血染色体的制作过程复杂,影响 因素众多,常常出现可分析分裂象小,染色体粗短,且分散度不良等现象,大大降低了染色 体核型分析的成功率,降低了其临床实用性,常规染色体分析对于<10m碱基的异常很难检出。 高分辨显带技术最终可以获得带纹550及以上的分裂相,可检出更多小片段染色体异常,建立 高分辨率染色体分析体系、提高小片段异常检出率,是临床细胞遗传关注的重要问题。因此 如何改进流程,提高染色体分带水平尤为重要。
技术实现要素:
[0005]
针对上述现有技术的不足,本发明的目的是提供一种外周血染色体g带的制作方法。采 用本发明的方法制作的外周血染色体呈长棒状,分散良好无交叉,显带清晰,带纹水平高。
[0006]
为实现上述目的,本发明采用如下技术方案:
[0007]
一种外周血染色体g带的制作方法,包括以下步骤:
[0008]
(1)将外周血0.3ml接种到外周血细胞培养基中培养66-72h;
[0009]
(2)向外周血细胞培养基中加入秋水仙素,终止培养;其中,每5ml外周血细胞培养基 中加入0.4ug/ml的秋水仙素;
[0010]
(3)收取外周血细胞悬液,调节细胞悬液的浓度为1.2-3.6
×
109/l;
[0011]
(4)将细胞悬液滴于玻片上,置于75℃烤箱烘干,烘干时间为3h;
[0012]
(5)烘干后的玻片采用浓度为0.01%的胰蛋白酶进行分带处理6-10min;
[0013]
(6)采用姬姆萨染料染色4min30s,即获得具有g显带的染色体标本。
[0014]
一些优选的实施方式中,步骤(1)中,所述外周血细胞培养基为小牛血清、pha外周血 培养液;培养于37℃二氧化碳培养箱。
[0015]
一些优选的实施方式中,步骤3)中,收取外周血细胞悬液的方法为:
[0016]
将培养物移至离心管中,以1500r/min离心5min,弃去上清液,沉淀物约留0.5~1ml;
[0017]
加入5ml低渗液至离心管中,用吸管吹打均匀制成细胞悬液,置34℃温箱内,以使红细 胞破坏,淋巴细胞膨胀,染色体分散;
[0018]
加入0.25ml固定液混匀预固定,以1500r/min离心5min;其中,固定液为甲醇和冰醋酸 按体积比3∶1混合而成;
[0019]
弃去上清液,加入固定液5ml,立即用吸管吹打细胞团块使之混匀,以1500r/min离心 5min;依上法固定3次;
[0020]
将固定后的沉淀物加新鲜固定液0.3~0.5ml,用吸管吹打混匀,制成细胞悬液。
[0021]
一些优选的实施方式中,将细胞悬液于5cm左右高度处滴于玻片上,以利于悬液均匀散 开。
[0022]
一些优选的实施方式中,将玻片置于75℃烤箱烘干,烘干时间为3h。
[0023]
本发明的有益效果:
[0024]
采用本发明方法制备的外周血染色体标本,与常规外周血染色体标本相比,其分裂相 更多,分裂相中染色体的长度更长,带纹更加清晰,分散度更加良好,便于分析;尤其对于 小片段异常染色体检出率更高,诊断结果更加可靠;采用本发明的外周血染色体g带用于外周 血染色体核型分析时,成功率显著提高,还降低了制作成本,具有广泛的应用前景。
附图说明
[0025]
图1:实施例1制备的外周血染色体在油镜下放大倍数
×
100的分裂相;
[0026]
图2:实施例1制备的外周血染色体排列图的分裂相;
[0027]
图3:对比例1制备的外周血染色体在油镜下放大倍数
×
100的分裂相;
[0028]
图4:对比例1制备的外周血染色体排列图的分裂相。
具体实施方式
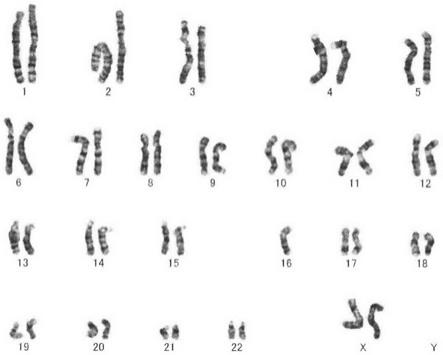
[0029]
正如背景技术所介绍的,由于外周血染色体的制作过程复杂,时间周期长,影响因素 多,经常出现染色体比较少,短、分散不好且有部分染色体交叉,显带也不理想等情况。基 于此,本发明提出了一种新的外周血染色体g带的制作方法,通过优化改进,增加染色体数量; 通过对收获方式、滴片方式进行改进,使染色后细胞大而圆润,染色体呈长棒状,分散良好 无交叉,显带清晰。
[0030]
有研究通过在传统的外周血培养基中加药物,在培养基中加入氟脱氧尿嘧啶核苷(fdu) 和尿嘧啶核苷(ur)使细胞分裂同步于s期,一段时间后加入胸腺嘧啶核苷(tdr)释放细胞,使 有丝分裂继续,最后在秋水仙素的作用下收获细胞。制出的g带背景更加清晰,形态及分散度 更加良好,分带水平较高。但是,由于在培养基中增加了药物后,增加了生产成
本。本发明 的改进思路不同于该研究,本发明仍采用传统的外周血细胞培养基,重点在于外周血染色体 制作方法的改进,这样不仅可以获得背景清晰的g带,分裂相多、形态及分散度优异的外周血 染色体,同时还降低了生产成本。
[0031]
秋水仙素在细胞收获过程中具有防止分裂中期微管(纺锤丝)形成或解聚的功能,此外 还能促使dna的复制和有关蛋白质合成,抑制细胞中微观形成,使细胞处于分裂中期。
[0032]
此外,秋水仙素也具有一定的细胞毒性。应用秋水仙素终止细胞培养,使其停留在中 期分裂相,是目前普遍采用的一种方法。但秋水仙素的处理浓度一直是争论的焦点,不同报 道的秋水仙素处理浓度相差很大,至今仍无一致的观点。秋水仙素能阻留中期分裂相,同时 也会使染色体浓缩,若秋水仙素的处理浓度过高,虽然分裂相会有所增加,但染色体长度反 而会缩短,导致带纹减少;若处理浓度过低,则会导致染色体分裂相减少。一般秋水仙素的 用法和用量遵循如下原则:秋水仙素浓度高,则处理时间短;浓度低,则处理时间延长。若 秋水仙素浓度过大,处理时间长,可导致染色体凝集和收缩变小,或发生异常分裂现象等情 况;而秋水仙素浓度过低,处理时间短,则染色体形态偏长或无中期分裂相。因此,在细胞 培养时一定要处理好秋水仙碱的量和作用时间,这是获得较好质量中期细胞分裂相的关键环 节。本研究发现,以终浓度为0.4μg/ml的秋水仙素处理细胞3h,可获得比较理想的染色体标 本;因此采用0.4μg/ml的秋水仙素作用3小时,终止细胞培养,取得了优异的效果,染色体 长度佳,结构紧密,分裂相多,显带成功率明显提高。采用秋水仙素终止细胞培养后,应用 低渗液处理细胞,可使染色体分散。低渗的原理是利用细胞内k+浓度远于细胞外k+浓度,使 淋巴细胞吸水膨胀,进而起到分散中期染色体的目的。低渗时间过短会导致细胞不易破裂, 反之则会造成细胞过早破裂和染色体丢失情况,且因染色体胀的太大导致染色体形态结构模 糊,影响后期染色效果。因此,需把握好低渗时间和低渗温度。目前多采用20-40min低渗, 可达到效果较好。也有研究发现,在短时间内完成低渗,不但可以获得较好中期细胞分裂相, 也可有效节约实验时间。然而,近期也有研究者尝试将秋水仙素处理和低渗同步处理,亦取 得了较好的实验效果。本实验结果对淋巴细胞低渗温度采用37℃下,以0.075mol/l的kcl进行 低渗,低渗30-50min,染色体的分散效果最佳。
[0033]
低渗后再采用固定液固定细胞,可以使带型清晰,若细胞固定不充分,会影响染色体 的显带质量。采用固定液进行固定中期淋巴细胞,使细胞脱水、蛋白变性、染色体稳定到适 当大小的关键步骤。本发明对细胞悬液的浊度进行了优化控制,用常用固定液为3体积的甲醇 和1体积的冰醋酸进行混合,固定液每次使用必须新鲜配制,否则将会形成酯类,形成背景较 为明显,从而影响制片效果,采用以1500r/min离心5min,来预固定1次,固定3次来实现固定 效果。其中预固定过程中预固定剂的剂量对中期染色体分散度的影响较大。本发明通过一次 预固定和三次固定,细胞得到了充分固定,更加有利于后续的染色操作。
[0034]
滴片效果影响中期染色体在玻片上的分散情况,直接影响染色体的分散效果。载玻片 的清洁度、悬液浓度、滴片时的高度等都直接影响染色体的分散效果。在滴片时,滴片以清 洗后预冷的滴片最宜,便于细胞滴在载玻片上有均匀的分散。悬液浓度应根据细胞量多少和 秋水仙碱作用时间来确定。本发明发现滴片时高度保持在5cm左右为宜,同时该高度不会造成 细胞和染色体的丢失及悬液飞溅污染其他标本。
[0035]
烤片可起到老化标本和蒸发残留于标本上固定剂的作用,经处理后的中期细胞有利于 胰蛋白酶消化液ph值的稳定性等。烤片的温度、时间与染色体形态和分带有关,温度
不宜过 高和烤片时间不宜过长。本研究表明75℃烤片3h最佳。
[0036]
胰酶消化固定于载玻片上的中期染色体是进行g显带的前提。胰酶4℃消化效果最好。 消化用胰酶浓度为0.01%。
[0037]
置于磷酸盐缓冲液(ph7.4),消化处理6min起,进行试染片,确定合适的消化时间。 将消化后的玻片置于生理盐水中终止胰酶的反应,后于添加有15%giemsa染液中进行染色, 染色深度不够可进行复染,以保证染色效果。提示可根据胰酶浓度,适当调整消化的时间, 以获得效果条带较好的染色体核型。本发明对胰蛋白酶的处理时间进行了优化,结果发现, 胰蛋白酶的处理时间为6-10分钟,磷酸盐缓冲液终止消化,染色体g显带的效果最优。
[0038]
为了使得本领域技术人员能够更加清楚地了解本技术的技术方案,以下将结合具体的 实施例详细说明本技术的技术方案。
[0039]
实施例1:外周血染色体g带的制作具体制作方法包括如下步骤:
[0040]
(1)准备培养基:将外周血培养基从冷冻冰箱取出,放37℃水浴箱温育,备用。
[0041]
(2)接种:每5ml外周血细胞培养基内外周血的接种量为0.5ml。
[0042]
(3)培养:在37℃的培养箱内进行培养,培养66-72h。
[0043]
(4)终止培养:收获前每瓶加入100μl的10μg/ml秋水仙素,再放入37℃的培养箱,终 止细胞分裂,使其停留在细胞中期,3h后收获。
[0044]
(5)收取外周血细胞培养物:
[0045]
a.离心:1500rpm/min,5分钟,弃上清液;
[0046]
b.低渗:加入预温至37℃的0.075m kcl低渗液5ml左右,轻轻混匀,置34℃,30分钟;
[0047]
c.预固定:加入0.25ml固定液轻轻混匀,1500rpm/min,5分钟。固定液配置比例:甲 醇和冰醋酸按3∶1体积混匀配成;
[0048]
d.第一次固定:离心后弃上清液,加入固定液约5ml,1500rpm/min,5分钟。
[0049]
e.第二次固定:1500rpm/min,5分钟,离心后弃上清液,加入固定液约5ml,混匀, 重复一次本步骤;
[0050]
f.离心1500rpm/min,5分钟,弃上清液,预留适量固定液配成细胞悬液,浓度1.2-3.6
ꢀ×
109/l,配好待用。
[0051]
(6)染色体标本制片:
[0052]
a滴片:
[0053]
a.取出在4℃冰箱中预冷的清洁玻片,对玻片进行标识,每标记完一块滴片板上的玻 片,就回顾一下是否编号准确,同时检查板上玻片的实验号和玻片放置的顺序,要做好一一 对应。
[0054]
b.用吸管吸取细胞悬液在5cm左右的高度滴于预冷的清洁玻片上。置于75℃的烤箱中 烘烤3小时。
[0055]
b染片:
[0056]
1)试剂准备
[0057]
a.0.01%胰酶配制,取0.02g的胰酶,加入200ml的pbs缓冲液中,充分混匀后,置于 水浴箱里标示胰酶的染缸中待用。
[0058]
b.将胰酶的染缸放在冰盒中,先置于-40℃冰箱中预冷20分钟。
[0059]
c.pbs缓冲液准备,取pbs缓冲液200ml置于水浴箱里标示pbs缓冲液的染缸中待用。
[0060]
d.姬姆萨染液配制,取11ml姬姆萨母液,加入200ml的pbs缓冲液中,充分混匀后,置 于水浴箱里标示染液的染缸中待用。
[0061]
e.蒸馏水准备,取200ml蒸馏水,加入到标示蒸馏水的染缸中待用。
[0062]
2)胰酶消化及染色:
[0063]
将玻片放入标示胰酶的染缸中消化6-10min,然后用pbs缓冲漂洗,立刻放入姬姆萨染 液中染色4分半钟。用蒸馏水冲洗。
[0064]
(7)显微镜下诊断,分析系统拍片:
[0065]
每批标本显带、染色完成后,镜下观察显带和染色效果,结果见图1、2。由图可以看 出,染色体的长度较长,形态清晰,带纹清晰可辨,分散度较好。
[0066]
对比例1:染色体g带的制作
[0067]
(1)准备培养基:将外周血培养基从冷冻冰箱取出,放37℃水浴箱温育,备用。
[0068]
(2)接种:每5ml外周血细胞培养基内外周血的接种量为0.5ml。
[0069]
(3)培养:在37℃的培养箱内进行培养,培养66-72h。
[0070]
(4)终止培养:收获前每瓶加入100μl的10μg/ml秋水仙素,再放入37℃的培养箱,终 止细胞分裂,使其停留在细胞中期,3h后收获。
[0071]
(5)收取外周血细胞培养物:
[0072]
a.离心:1500rpm/min,5分钟,弃上清液;
[0073]
b.低渗:加入预温至37℃的0.075m kcl低渗液5ml左右,轻轻混匀,置35℃,20分钟;
[0074]
c.预固定:加入0.25ml固定液轻轻混匀,1500rpm/min,5分钟。固定液配置比例:甲 醇和冰醋酸按3∶1体积混匀配成;
[0075]
d.第一次固定:离心后弃上清液,加入固定液约5ml,1500rpm/min,5分钟。
[0076]
e.第二次固定:1500rpm/min,5分钟,离心后弃上清液,加入固定液约5ml,混匀, 重复一次本步骤;
[0077]
f.离心1500rpm/min,5分钟,弃上清液,预留适量固定液配成细胞悬液,浓度1.2-3.6
ꢀ×
109/l,配好待用。
[0078]
(6)染色体标本制片:
[0079]
a滴片:
[0080]
a.取出在清洁玻片,对玻片进行标识,每标记完一块滴片板上的玻片,就回顾一下是 否编号准确,同时检查板上玻片的实验号和玻片放置的顺序,要做好一一对应。
[0081]
b.用吸管吸取细胞悬液在5cm左右的高度滴于预冷的清洁玻片上。置于75℃的烤箱中 烘烤3小时。
[0082]
b染片:
[0083]
1)试剂准备
[0084]
a.0.01%胰酶配制,取0.02g的胰酶,加入200ml的pbs缓冲液中,充分混匀后,置于 水浴箱里标示胰酶的染缸中待用。
[0085]
b.将胰酶的染缸先置于冰盒中预冷20分钟。
[0086]
c.pbs缓冲液准备,取pbs缓冲液200ml置于水浴箱里标示pbs缓冲液的染缸中待用。
[0087]
d.姬姆萨染液配制,取11ml姬姆萨母液,加入200ml的pbs缓冲液中,充分混匀后,置 于水浴箱里标示染液的染缸中待用。
[0088]
e.蒸馏水准备,取200ml蒸馏水,加入到标示蒸馏水的染缸中待用。
[0089]
2)胰酶消化及染色:
[0090]
将玻片放入标示胰酶的染缸中消化6-10min,然后用pbs缓冲漂洗,立刻放入姬姆萨染 液中染色4分半钟。用蒸馏水冲洗。
[0091]
(7)显微镜下诊断,分析系统拍片:
[0092]
镜下观察显带和染色效果,结果见图3,4。由图可以看出,染色体短小,边缘毛糙, 分散度较差,带纹较模糊。
[0093]
由实施例1和对比例1可以看出,外周血细胞的培养时间、低渗温度和时间及滴片后的 烤片时间,会直接影响外周血染色体制备的质量。利用本发明的方法(实施例1),图1-2和3-4 对比,能增加外周血染色体分裂相数目,染色体长度增加,形态更加良好,带纹更加清晰可 辨,制取的g带稳定性更好,能更好的检出异常染色体,诊断结果更加可靠。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1