复方氨基酸注射液中微量半胱氨酸含量的检测方法与流程
本发明涉及一种复方氨基酸注射液中半胱氨酸含量的检测方法,本发明尤其是一种应用高效液相色谱法复方氨基酸注射液中微量半胱氨酸含量的检测方法。
背景技术:
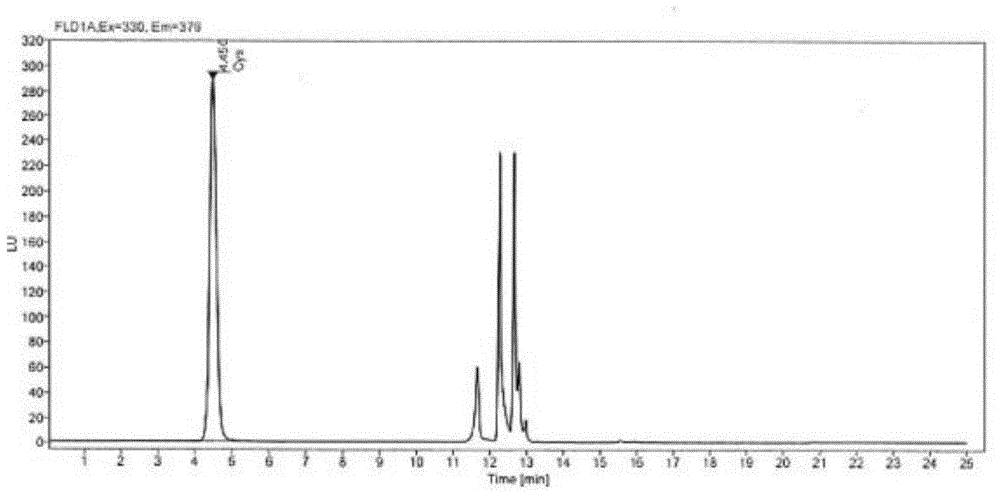
:复方氨基酸注射液为肠外营养药,种类繁多,处方各异,据不完全统计,含有胱氨酸、半胱氨酸、乙酰半胱氨酸的复方氨基酸注射液有18个,经本课题组研究发现复方氨基酸注射液中胱氨酸和乙酰半胱氨酸会降解生成微量的半胱氨酸。因此,在复方氨基酸注射液中,半胱氨酸的含量可以反映产品的质量。半胱氨酸的极性较大,使用c18色谱柱,保留很弱,难分离,离子对色谱法通过向流动相加入离子对试剂,增强了c18色谱柱对极性化合物的保留,但存在保留时间不重现、色谱柱寿命短的问题;由于复方氨基酸注射液处方中胱氨酸、半胱氨酸、乙酰半胱氨酸的规格较小且共存氨基酸较多,常用的柱前或柱后衍生氨基酸分析方法由于灵敏度不够或者半胱氨酸与共存氨基酸的分离度欠佳,无法对半胱氨酸进行准确定量。现有技术(cn106950306a)中有报道采用n-(1-芘基)马来酰亚胺作为衍生剂对半胱氨酸进行衍生,采用高效液相色谱测定复方氨基酸注射液中半胱氨酸的含量(处方中含有半胱氨酸),该技术中采用的是紫外检测器,对于处方中不含半胱氨酸,如含有胱氨酸和乙酰半胱氨酸的复方氨基酸注射液中微量的半胱氨酸含量的测定,方法的灵敏度不够。技术实现要素:本发明的目的是克服现有技术中存在的不足,提供一种专属性好、灵敏度佳且衍生反应速度快的复方氨基酸注射液中微量半胱氨酸含量的检测方法。按照本发明提供的技术方案,所述复方氨基酸注射液中微量半胱氨酸含量的检测方法包括以下步骤:步骤一、半胱氨酸加水稀释成浓度为0.0639~20.04mg/l后作为对照品溶液;步骤二、复方氨基酸注射液加水进行稀释后作为供试品溶液;步骤三、采用浓度为0.8~2.6mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液对所述对照品溶液和供试品溶液中半胱氨酸分别进行衍生,对照品溶液以及供试品溶液与n-(1-芘基)马来酰亚胺乙腈溶液反应的体积比均为1:3,衍生完成后加入盐酸溶液涡旋混匀;步骤四、确定以下色谱条件:色谱柱采用250×4.6mm、5μm的十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm的磷酸二氢钠缓冲液,流动相b为乙腈;线性梯度洗脱条件为:0~8分钟:60%~56%流动相a、40%~44%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:60%~56%流动相a、40%~44%流动相b;流速设定为0.9~1.1ml/min;柱温设定为33~37℃;激发波长设定为330nm;发射波长设定为376nm;进样器温度设定为4℃;进样体积设定为10μl;步骤五、将所述步骤三得到的衍生后的对照品溶液和供试品溶液采用步骤四中条件进行高效液相色谱分析,根据衍生后的对照品溶液和供试品溶液的液相色谱图,采用外标法得到复方氨基酸注射液中半胱氨酸的含量。作为优选,步骤一中,半胱氨酸加水稀释成浓度为20.04mg/l后作为对照品溶液。作为优选,步骤二中,复方氨基酸注射液稀释倍数为10倍。作为优选,步骤三中,衍生时间为10min~30min。作为优选,步骤三中,在衍生化反应后加入的盐酸溶液的浓度为0.2mol/l,盐酸溶液体积与衍生化后对照品溶液的体积比为1:10,盐酸溶液体积与衍生化后供试品溶液的体积比为1:10。作为优选,步骤四中,所用柱温为35℃。作为优选,步骤四中,所用流速为1.0ml/min。作为优选,步骤四中,所述线性梯度洗脱如下:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b。作为优选,步骤四中,准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至2.80~3.20,用0.45μm滤膜过滤,得到所述的磷酸二氢钠缓冲液。本发明提供的方法能够实现复方氨基酸注射液中微量半胱氨酸含量的准确测定,且专属性好、精密度好、回收率好、灵敏度高。与现有技术相比,本发明优势如下:采用荧光检测器,灵敏度更高,专属性更好;操作简单,衍生反应在室温下10min内完成;本发明衍生反应结束后加入盐酸,对照品溶液和供试品溶液中半胱氨酸衍生化产物稳定性较好,可在4℃条件下稳定数日。附图说明图1是实施例1中对照品工作溶液的色谱图。图2是实施例1中供试品溶液的色谱图。图3是实施例2中供试品溶液的色谱图。图4是实施例3中供试品溶液的色谱图。图5是实施例4中供试品溶液的色谱图。图6是实施例5中供试品溶液的色谱图。图7是实施例6中供试品溶液的色谱图。图8是实施例7中供试品溶液的色谱图。图9是实施例8中供试品溶液的色谱图。图10是实施例9中供试品溶液的色谱图。图11是实施例10中供试品溶液的色谱图。具体实施方式下面结合具体实施例对本发明作进一步说明。以下实施例使用的色谱柱是十八烷基硅烷键合硅胶c18色谱柱,产品参数为5μm,250mm×4.6mm。实施例1一种复方氨基酸注射液(处方中含胱氨酸)中半胱氨酸含量的检测方法包括以下步骤:步骤一、半胱氨酸加水稀释成浓度为20.040mg/l后作为对照品溶液;步骤二、复方氨基酸注射液加水进行稀释10倍后作为供试品溶液;步骤三、采用浓度为2.6mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述对照品溶液50μl和步骤二所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤四、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.0ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤五、将所述步骤三得到的衍生后的对照品溶液和供试品溶液采用步骤四中条件进行高效液相色谱分析,根据衍生后的对照品溶液和供试品溶液的液相色谱图,采用外标法得到复方氨基酸注射液中半胱氨酸的含量。检测结果:对照品工作溶液和供试品溶液的保留时间一致。供试品溶液中半胱氨酸衍生物与相邻色谱峰的分离度为7.93,见表1。表1样品保留时间(min)分离度对照品工作溶液4.45028.163供试品溶液4.4659.770实施例2一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.0ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表2。表2样品保留时间(min)分离度供试品溶液4.548.98实施例3一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.0ml/min;柱温:33℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表3。表3样品保留时间(min)分离度供试品溶液4.5568.97实施例4一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.0ml/min;柱温:37℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表4。表4实施例5一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:0.9ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表5。表5样品保留时间(min)分离度供试品溶液4.8679.01实施例6一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.1ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表6。表6样品保留时间(min)分离度供试品溶液4.2148.97实施例7一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:60%流动相a、40%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:60%流动相a、40%流动相b;流速:1.0ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表7。表7样品保留时间(min)分离度供试品溶液5.0348.46实施例8一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.0,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:56%流动相a、44%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:56%流动相a、44%流动相b;流速:1.0ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表8。表8样品保留时间(min)分离度供试品溶液4.1447.94实施例9一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至2.8,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.0ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表9。表9样品保留时间(min)分离度供试品溶液4.3708.53实施例10一种应用高效液相色谱法检测复方氨基酸注射液(处方中含乙酰半胱氨酸)中半胱氨酸含量的检测方法,包括以下步骤:步骤一、复方氨基酸注射液(添加半胱氨酸浓度64mg/l)加水进行稀释10倍后作为供试品溶液;步骤二、采用浓度为0.8mmol/l的n-(1-芘基)马来酰亚胺乙腈溶液150μl分别与步骤一所述供试品溶液50μl混合进行衍生,衍生完成后加入0.2mol/l盐酸溶液20μl涡旋混匀;步骤三、色谱条件为:色谱柱:(250×4.6mm,5μm)十八烷基硅烷键合硅胶c18色谱柱;流动相:流动相a为10mm磷酸二氢钠缓冲液(准确称取磷酸二氢钠1.56g至1000ml水中,溶解后,加磷酸调ph至3.2,用0.45μm滤膜过滤);流动相b为乙腈,进行线性梯度洗脱,线性梯度洗脱条件为:0~8分钟:58%流动相a、42%流动相b;9~17分钟:10%流动相a、90%流动相b;18分钟以后:58%流动相a、42%流动相b;流速:1.0ml/min;柱温:35℃;激发波长:330nm发射波长:376nm进样器温度:4℃进样体积:10μl步骤四、将所述步骤二得到的衍生后的供试品溶液采用步骤三中条件进行高效液相色谱分析,记下半胱氨酸衍生产物的色谱峰位置和与相邻色谱峰的分离度见表10。表10样品保留时间(min)分离度供试品溶液4.4228.82本发明方法的线性按实施例2的色谱条件对线性关系进行测定,分别配制浓度为0.0639μg/ml、0.6386μg/ml、1.5965μg/ml、3.1930μg/ml、6.3860μg/ml、9.5790μg/ml、12.7720μg/ml的对照品溶液,以峰面积与其对应浓度进行线性回归,结果表明,对照品溶液中半胱氨酸浓度为0.0639mg/l~12.7720mg/l之间,相当于供试品中半胱氨酸浓度为0.6390mg/l~127.7200mg/l时,以峰面积与其对应的浓度进行回归计算,回归方程为y=105.4809x+2.0419,相关系数r=0.9999。本发明方法的准确度和精密度按实施例1的色谱条件对本发明方法的准确度和精密度进行试验,分别向不含有半胱氨酸的样品中添加半胱氨酸,设计3种不同浓度,每种浓度分别制备3份供试品溶液进行测定,用9份样品的测定结果进行评价,测定结果见表10。表10说明:n-乙酰半胱氨酸对照品中含有微量半胱氨酸,配制半胱氨酸空白供试品中加入了n-乙酰半胱氨酸对照品,因此会引入微量半胱氨酸,在计算回收率时应将这部分半胱氨酸扣除。从表10中可以看出,供试品中半胱氨酸浓度在3.1740mg/l~9.7360mg/l范围内,平均回收率为100.2%,rsd为1.3%。本发明方法的定量限按实施例1的色谱条件对本发明方法的定量限进行试验,结果表明,在信噪比不小于10的条件下,本发明检测方法的定量限为供试品中半胱氨酸浓度为1.8870mg/l。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1