一种鸡蛋中氟虫腈及其代谢物的净化和检测方法与流程
本发明涉及氟虫腈及其代谢物检测的
技术领域:
,具体涉及一种鸡蛋中氟虫腈及其代谢物的净化和检测方法。
背景技术:
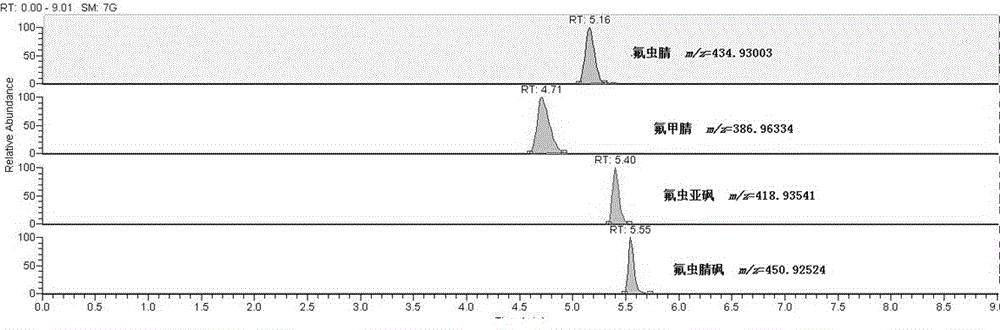
:氟虫腈是一种新型苯基吡唑类杀虫剂,广泛用于农业害虫防治。苯基吡唑是一种神经毒性物质,能与昆虫的离子型γ-氨基丁酸受体(gabar)结合,干扰氯离子的通路,在亚致死剂量时具有某些兴奋效应,而在高剂量时会导致死亡。此外,氟虫腈可以通过氧化、还原、水解和光解形成三种主要代谢产物:氟虫腈砜、氟虫腈亚砜和氟甲腈。据动物实验研究表明,长期摄取受氟虫腈污染的食物,动物的肝脏、肾和甲状腺功能会受到损伤。2017年7月,比利时有关部门通过欧盟食品饲料类快速预警系统(rasff)发现产自荷兰的鸡蛋中检测出氟虫腈,同年8月,问题鸡蛋波及欧洲16个国家、韩国等国家及中国香港、台湾地区,使“毒鸡蛋”事件成为世界范围内的重大食品安全事件,引起公众广泛关注。欧盟相关标准规定氟虫腈在蛋中的最大残留限量为0.005mg/kg,我国食品安全国家标准gb2763-2016《食品安全国家标准食品中农药最大残留限量》中仅明确了氟虫腈及其代谢物在蔬菜、水果、油料、谷物等植物源性食品的限量(玉米为0.1mg/kg,其他为0.02mg/kg),蛋类中的限量未明确。有研究表明,鸡蛋经过加工烹饪后,氟虫腈及其代谢物的含量不会发生明显变化,因此,鸡蛋中氟虫腈及其代谢物的检测尤为重要。目前,有关鸡蛋中氟虫腈及其代谢物的检测标准仅有gb23200.115-2018食品安全国家标准鸡蛋中氟虫腈及其代谢物残留量的测定采用的是液相色谱-质谱联用法,该标准使用quechers方法净化,虽然简单、快速,但其原理是通过去除杂质来达到净化效果,所以方法的定量限较高(0.005mg/kg)。和该标准的净化方法类似的有公开号为cn107505421a(名称为一种鸡蛋中氟虫腈及其代谢物残留的快速提取及净化方法)和公开号为cn107860841a(名称为一种检测鸡蛋中氟虫腈及其代谢物的方法氟虫腈及其代谢物)的发明专利,同样方法的定量限较高(均大于0.5μg/kg)。而其他的行业内的检测手段还存在一定的不足,国家知识产权局于2019年5月14日公开的一件公开号为cn109752531a,名称为一种检测鸡蛋中氟虫腈的试剂盒及其检测方法的发明专利,该专利使用的酶联免疫吸附测定法灵敏度低,而且会出现假阳性结果;授权号为cn107037149b(名称为一种鸡蛋中氟虫腈及其代谢物残留量测定的方法)和公开号为cn107917974a(名称为一种鸡蛋中氟虫腈及其代谢物残留量的检测方法)的发明专利,使用的是固相萃取柱净化,操作步骤复杂且时间较长;2019年1月18日公开的公开号为cn109239248a(名称为一种蛋及蛋制品中氟虫腈的lc-q-tof检测方法)的发明专利,该方法仅只检测氟虫腈,不包括其代谢物(氟甲腈、氟虫腈亚砜和氟虫腈砜),且使用的净化方法是超临界co2萃取方法,需要用到萃取釜装置,仪器投入大,过程复杂、操作要求高。为了去除基质干扰,提高方法灵敏度,常用的氟虫腈及其代谢物的前处理方法包括固相萃取法(spe)和quechers方法。然而,传统的固相萃取法操作繁琐,成本高,耗费时间长;而quechers方法虽然简单、快速,但其原理是通过去除杂质来达到净化效果,所以对于目标化合物的回收率较低。本申请人发现现有技术至少存在以下技术问题:1、现有技术中的鸡蛋中氟虫腈及其代谢物的净化和检测方法,其定量限较高,检测灵敏度不够,检测精确度不高;2、现有技术中的鸡蛋中氟虫腈及其代谢物的净化和检测方法,使用了固相萃取柱净化,操作步骤复杂且时间较长;3、现有技术中的鸡蛋中氟虫腈及其代谢物的净化和检测方法,较多使用的仪器是低分辨质谱,低分辨质谱由于分辨率低,有时不能有效识别目标物与杂质,导致结果可靠性低。技术实现要素:本发明的目的在于提供一种鸡蛋中氟虫腈及其代谢物的净化和检测方法,以解决现有技术中存在的现有技术中现有技术中的鸡蛋中氟虫腈及其代谢物的净化和检测方法,其定量限较高,检测灵敏度不够,检测精确度不高的技术问题。本发明提供的诸多技术方案中的优选技术方案所能产生的诸多技术效果详见下文阐述。为实现上述目的,本发明提供了以下技术方案:本发明提供的一种鸡蛋中氟虫腈及其代谢物的净化和检测方法,包括下述步骤:(1)制备磁性纳米材料a1、将柠檬酸钠、乙酸钠和fecl3·6h2o通过乙二醇溶解后在高压反应釜中反应生成fe3o4纳米颗粒,经磁分离、洗涤、冷冻干燥后,得fe3o4纳米粒子;此步骤中,fecl3被乙二醇还原得到fe3o4颗粒,乙酸钠起到防止fe3o4颗粒团聚的作用,柠檬酸钠则可以对四氧化三铁颗粒进行表面处理;a2、将步骤a1中制备的fe3o4纳米粒子经过盐酸超声处理、洗涤后,将洗涤后的fe3o4纳米粒子、甲醇、水和氨水溶液加入反应容器中,向反应容器中滴加teos,反应后生成的纳米粒子,经磁分离、洗涤后,得fe3o4@nsio2纳米粒子;a3、将步骤a2中合成的fe3o4@nsio2纳米粒子、水、无水乙醇、ctab、氨水溶液加入反应容器中进行超声分散,超声分散后在搅拌的状态下滴加teos进行反应,反应生成的纳米粒子,经洗涤、冷冻干燥,得fe3o4@nsio2@msio2纳米粒子;a4、将步骤a3中合成的fe3o4@nsio2@msio2纳米粒子分散在tris溶液中,加入多巴胺,将反应生成的纳米粒子进行磁分离后,经洗涤、冷冻干燥,得fe3o4@nsio2@msio2-pda磁性纳米材料;(2)样品前处理b1、新鲜鸡蛋去壳后搅拌混匀,然后将搅拌混匀的鸡蛋液通过乙腈进行超声提取,最后进行磁性固相萃取净化,得到含氟虫腈及其代谢物的稀释溶液;b2、向步骤b1中所得的稀释溶液中加入步骤(1)中制备的磁性纳米材料,使稀释溶液中的氟虫腈及其代谢物被吸附到磁性纳米材料上;b3、用磁铁将吸附了氟虫腈及其代谢物的磁性纳米材料与溶液进行磁分离,弃去上清液,进行洗涤;再次进行磁分离,弃去上清液;b4、使用乙腈将步骤b3中吸附了氟虫腈及其代谢物的磁性纳米材料上的氟虫腈及其代谢物的洗脱下来,然后进行磁分离,将磁性纳米材料分离出来,保留上清液;b5、将步骤b4中的上清液经滤膜过滤处理,得待测样品;(3)采用超高效液相色谱-q-exactive高分辨质谱进行检测分析。进一步的,所述步骤(1)a1中,在高压反应釜中反应是在温度为190℃-210℃反应11h-13h;所述fecl3·6h2o和乙二醇间的比例为1:50~200;其中,所述fecl3·6h2o以g计,所述乙二醇以ml计。进一步的,所述步骤(1)a2中,所述fe3o4纳米粒子和teos间的比例为1:2~10;其中,所述fe3o4纳米粒子以g计,所述teos以ml计。进一步的,所述步骤(1)a3中,所述fe3o4@nsio2纳米粒子、ctab和teos间的比例为1:1~6:2~10;其中,所述fe3o4@nsio2纳米粒子和ctab均以g计,所述teos以ml计。进一步的,所述步骤(1)a4中,所述fe3o4@nsio2@msio2磁性纳米粒子与多巴胺间的重量比为1:1~5。进一步的,所述步骤(2)b1中,通过乙腈进行超声提取和进行磁性固相萃取净化具体为:称取1~5g搅拌混匀的鸡蛋液加入50ml离心管中,加入5~20ml乙腈,涡旋混匀1min,然后超声提取10~20min;加入0.5~2g氯化钠和1~5g无水硫酸镁,在转速为10000~15000r/min离心5~10min,接着于-18℃放置20~30min;移取1~5ml上清液于100ml离心管中,加入10~50ml水稀释,最后进行磁性固相萃取净化,磁性固相萃取净化时,磁性吸附剂用量为5~40mg,磁性吸附时间为1~15min,洗脱溶剂为乙腈,洗脱时间为1~15min,磁性固相萃取净化完成后,得到含氟虫腈及其代谢物的稀释溶液。进一步的,所述步骤(3)中,超高效液相色谱的条件为:dionexultimate3000超高效液相色谱仪配备wps-3000trs自动进样器、tcc-3000rs柱温箱和hpg-3400rs二元泵;色谱柱:accucoreaq(2.1×150mm,2.6μm粒径);流速:0.2~0.4ml/min;柱温:30-45℃;进样量:2~10μl;流动相由水(流动相a)和乙腈(流动相b)组成;梯度洗脱条件如下:0-3min,60%b;3-3.5min,60%b-90%b;3.5-4.5min,90%b;4.5-6min,90%b-60%b;6-9min,10%b。进一步的,所述超高效液相色谱的条件中,所述进样量为5μl;所述的柱温40℃。进一步的,所述步骤(3)中,高分辨质谱的条件为:鞘气流量:35个任意单位;辅助气流量:15个任意单位;尾吹气流量:1个任意单位;喷雾电压:1.5-3kv(负离子模式);扫描范围:m/z为200-600;毛细管温度:350℃;s透镜rf:50个任意单位;辅助气温度:350℃;一级质谱全扫描分辨率:r=70000;c-trap最大容量(targetagc):3e6;c-trap最长等待时间:200ms;数据依赖二级全扫描(dd-ms2)分辨率:r=17500;c-trap最大容量(targetagc):5e4;c-trap最长等待时间:50ms;动态排除:10s。进一步的,高分辨质谱的条件中,所述的喷雾电压为2.5kv。基于上述技术方案,本实施例至少可以产生如下技术效果:(1)本发明提供的鸡蛋中氟虫腈及其代谢物的净化和检测方法,制备出的磁性纳米材料为聚多巴胺功能化的磁性介孔二氧化硅纳米材料(fe3o4@nsio2@msio2-pda),多巴胺作为纳米材料的表面改性剂,可以通过自聚合反应形成聚多巴胺薄膜,应用于磁性介孔二氧化硅纳米材料的表面修饰和改性。将多巴胺与磁性介孔二氧化硅纳米材料结合,发挥其对氟虫腈及其代谢物潜在的吸附能力,将其应用到鸡蛋中氟虫腈及其代谢物检测,具有分析简便、快速,具有较高的灵敏度和精确度,重现性较好,定量准确且回收率较高,能够准确分析鸡蛋中氟虫腈及其代谢物的含量的优势。(2)本发明提供的鸡蛋中氟虫腈及其代谢物的净化和检测方法,是应用磁性介孔二氧化硅纳米材料(fe3o4@nsio2@msio2-pda)对氟虫腈及其代谢物的吸附能力,将其吸附分离后,再利用orbitrap高分辨率质谱检测技术进行检测,其具有更高的分辨率、准确的定性定量的优点,结果可靠性高;该检测方法具有分析简便、快速,具有较高的灵敏度和精确度,重现性较好,定量准确且回收率较高,能够准确分析鸡蛋中氟虫腈及其代谢物的含量的优势。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1是实施例1中空白鸡蛋样中添加氟虫腈及其代谢物的提取离子色谱图。具体实施方式为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。实施例1:一种鸡蛋中氟虫腈及其代谢物的净化和检测方法,包括下述步骤:(1)制备磁性纳米材料a1、取0.72g柠檬酸钠、4.8g乙酸钠和3.0gfecl3·6h2o,用300ml乙二醇溶解后加入高压反应釜中,在200℃反应12h;将反应生成的fe3o4纳米颗粒在外加磁场的作用下磁分离(通过外置磁铁进行溶液和磁性纳米颗粒的分离),经蒸馏水洗涤2次,无水乙醇洗涤1次,-50℃冷冻干燥12h,得fe3o4纳米粒子;a2、称取步骤a1中制备的0.3g的fe3o4纳米粒子加入反应容器内,加入150ml盐酸(0.1m)超声15min;然后用蒸馏水洗涤4次;向反应容器内加入240ml甲醇、60ml水和3.0ml氨水溶液,然后在不断搅拌下逐滴加入1.5ml的teos,在室温下反应24h;在外加磁场的作用下磁分离,经无水乙醇和蒸馏水各洗涤3次;得fe3o4@nsio2纳米粒子;取少量fe3o4@nsio2纳米粒子于-50℃冷冻干燥12h,用于表征;a3、将0.3g步骤a2中合成的fe3o4@nsio2粒子中加入240ml水、180ml无水乙醇、0.9gctab和3.3ml氨水溶液超声分散15min,然后不断搅拌下逐滴加入1.5ml的teos,室温下反应24h;用盐酸-乙醇溶液(5:95,v/v)洗去残留的ctab,经无水乙醇和水各洗涤3次,-50℃冷冻干燥12h,得fe3o4@nsio2@msio2纳米粒子;a4、称取0.5g步骤a3中合成的fe3o4@nsio2@msio2磁性纳米粒子,将其分散在150ml的tris溶液(10mm,ph8.5)中,加入2g多巴胺,室温机械搅拌反应多少时间生成磁性纳米材料,将生成的磁性纳米材料进行磁分离后,经蒸馏水洗涤三次,-50℃冷冻干燥12h,即得到fe3o4@nsio2@msio2-pda磁性纳米材料;(2)样品前处理b1、新鲜鸡蛋去壳后搅拌混匀,然后将搅拌混匀的鸡蛋液通过乙腈进行超声提取,最后进行磁性固相萃取净化,得到含氟虫腈及其代谢物的稀释溶液;通过乙腈进行超声提取和进行磁性固相萃取净化具体为:称取5g搅拌混匀的鸡蛋液加入50ml聚四氟乙烯离心管中,加入10ml乙腈,涡旋混匀1min,然后超声提取10min;加入1g氯化钠和3g无水硫酸镁,在转速为10000r/min离心5min,接着于-18℃放置20min,析出上清液;移取5ml上清液于100ml聚四氟乙烯离心管中,加入50ml水稀释;最后进行磁性固相萃取净化,磁性固相萃取净化时,磁性吸附剂用量为30mg,磁性吸附时间为5min,洗脱溶剂为乙腈,洗脱时间为5min,磁性固相萃取净化完成后,得到含氟虫腈及其代谢物的稀释溶液;b2、向步骤b1中所得的稀释溶液中加入30mg实施例1中制备的磁性纳米材料,采用涡旋混合仪涡旋混合5min,使稀释溶液中的氟虫腈及其代谢物被吸附到磁性纳米材料上;b3、用磁铁将吸附了氟虫腈及其代谢物的磁性纳米材料与溶液进行磁分离,弃去上清液,再用蒸馏水洗涤两次;再次进行磁分离,弃去上清液;b4、使用1ml乙腈将步骤b3中吸附了氟虫腈及其代谢物的磁性纳米材料上的氟虫腈及其代谢物的洗脱下来,然后进行磁分离,将磁性纳米材料分离出来,保留上清液;b5、吸取步骤b4中的上清液0.5ml于离心管中,加入0.5ml水,混匀后经0.22μm滤膜过滤,得待测样品;(3)采用超高效液相色谱-q-exactive高分辨质谱将步骤(2)中所得的待测样品进行检测分析。超高效液相色谱的条件为:dionexultimate3000超高效液相色谱仪配备wps-3000trs自动进样器、tcc-3000rs柱温箱和hpg-3400rs二元泵;色谱柱:accucoreaq(2.1×150mm,2.6μm粒径);流速:0.3ml/min;柱温:40℃;进样量:5μl;流动相由水(流动相a)和乙腈(流动相b)组成;梯度洗脱条件如下:0-3min,60%b;3-3.5min,60%b-90%b;3.5-4.5min,90%b;4.5-6min,90%b-60%b;6-9min,10%b;高分辨质谱的条件为:鞘气流量:35个任意单位;辅助气流量:15个任意单位;尾吹气流量:1个任意单位;喷雾电压:2.50kv(负离子模式);扫描范围:m/z为200-600;毛细管温度:350℃;s透镜rf:50个任意单位;辅助气温度:350℃;一级质谱全扫描分辨率:r=70000;c-trap最大容量(targetagc):3e6;c-trap最长等待时间:200ms;数据依赖二级全扫描(dd-ms2)分辨率:r=17500;c-trap最大容量(targetagc):5e4;c-trap最长等待时间:50ms;动态排除:10s。待测化合物的信息和质谱参数见下表1所示:表1待测化合物的q-exactive高分辨质谱参数1.1线性、检出限和定量限:取空白鸡蛋样品,加入适量的氟虫腈及其代谢物标准工作溶液,按照步骤(2)的方法所述进行样品前处理,制备成标准样品,使6个浓度点的基质匹配校准曲线浓度范围为0.01-10μg/kg,再依次注入超高效液相色谱-q-exactive高分辨质谱进行分析,每个浓度重复测定三次,以氟虫腈及其代谢物的母离子的峰面积对浓度绘制标准曲线,结果如下表2所示:采用空白鸡蛋样品中添加氟虫腈及其代谢物的方法,按照步骤(2)的样品前处理方法和步骤(3)的仪器条件进行处理和检测,以3倍信噪比和10倍信噪比分别对应的氟虫腈及其代谢物的浓度计算检出限(lod)和定量限(loq)结果如下表2所示:表2鸡蛋中氟虫腈及其代谢物的线性,检出限和定量限从上表2可以看出,氟虫腈及其代谢物在0.01-10μg/kg浓度范围内线性关系良好,r2>0.997。本实施例4的检测方法的检出限和定量限分别为0.002-0.005μg/kg和0.006-0.015μg/kg。1.2准确度和精密度为了评价实施例1的检测方法的准确度和精密度,在空白鸡蛋样品中添加已知量的氟虫腈及其代谢物的混合标准工作溶液进行添加回收率实验,三个添加浓度分别为0.0100、0.100和1.00μg/kg,图1为空白鸡蛋样品中添加0.1μg/kg的氟虫腈及其代谢物的提取离子色谱图。对于日内精密度,空白鸡蛋加标样品每个浓度重复5次,计算rsd;对于日间精密度,空白鸡蛋加标样品每个浓度重复3次,连续测定3天,计算rsd,结果见表3,氟虫腈及其代谢物的回收率在80.6-95.3%之间。日内和日间精密度分别为2.2-6.7%和4.4-10.7%。表3鸡蛋样品中添加氟虫腈及其代谢物的回收率,日内精密度和日间精密度1.3应用实施例1中的检测方法进行样品分析从超市中购买了20个鸡蛋样品,并应用实施例1中的检测方法对这些样品中的氟虫腈及其代谢物进行分析,样品检测结果如下表4所示:表4样品检测结果氟虫腈(μg/kg)氟甲腈(μg/kg)氟虫腈亚砜(μg/kg)氟虫腈砜(μg/kg)1未检出未检出未检出未检出2未检出未检出未检出未检出3未检出未检出未检出未检出4未检出未检出未检出未检出5未检出未检出未检出未检出6未检出未检出未检出0.5887未检出未检出未检出未检出8未检出未检出未检出未检出9未检出未检出未检出未检出10未检出未检出未检出1.8511未检出未检出未检出未检出12未检出未检出未检出2.1413未检出未检出未检出未检出14未检出未检出未检出未检出15未检出未检出未检出未检出16未检出未检出未检出未检出17未检出未检出未检出未检出18未检出未检出未检出未检出19未检出未检出未检出未检出20未检出未检出未检出未检出由上表4可以看出,氟虫腈、氟甲腈和氟虫腈亚砜均未检测到,有3个样品(样品6、样品10和样品12)中检测出了氟虫腈砜,浓度分别为0.588μg/kg、1.85μg/kg和2.14μg/kg,低于欧盟规定的最高限量(0.005mg/kg),检测灵敏度和检测精确度均高于现有技术中的检测方法。实施例2:一种鸡蛋中氟虫腈及其代谢物的净化和检测方法,包括下述步骤:(1)制备磁性纳米材料a1、取0.72g柠檬酸钠、4.8g乙酸钠和3.0gfecl3·6h2o,用150ml乙二醇溶解后加入高压反应釜中,在190℃反应13h;将反应生成的fe3o4纳米颗粒在外加磁场的作用下磁分离,经蒸馏水洗涤2次,无水乙醇洗涤1次,-50℃冷冻干燥12h,得fe3o4纳米粒子;a2、称取步骤a1中制备的0.3g的fe3o4纳米粒子加入反应容器内,加入150ml盐酸(0.1m)超声15min;然后用蒸馏水洗涤4次;向反应容器内加入240ml甲醇、60ml水和3.0ml氨水溶液,然后在不断搅拌下逐滴加入0.6ml的teos,在室温下反应24h;在外加磁场的作用下磁分离,经无水乙醇和蒸馏水各洗涤3次;得fe3o4@nsio2纳米粒子;取少量fe3o4@nsio2纳米粒子于-50℃冷冻干燥12h,用于表征;a3、将0.3g步骤a2中合成的fe3o4@nsio2粒子中加入240ml水、180ml无水乙醇、0.3gctab和3.3ml氨水溶液超声分散20min,然后不断搅拌下逐滴加入0.6ml的teos,室温下反应26h;用盐酸-乙醇溶液(5:95,v/v)洗去残留的ctab,经无水乙醇和水各洗涤3次,-50℃冷冻干燥12h,得fe3o4@nsio2@msio2纳米粒子;a4、称取0.5g步骤a3中合成的fe3o4@nsio2@msio2磁性纳米粒子,将其分散在150ml的tris溶液(10mm,ph8.5)中,加入0.5g多巴胺,室温机械搅拌反应多少时间生成磁性纳米材料,将生成的磁性纳米材料进行磁分离后,经蒸馏水洗涤三次,-50℃冷冻干燥12h,即得到fe3o4@nsio2@msio2-pda磁性纳米材料。(2)样品前处理b1、新鲜鸡蛋去壳后搅拌混匀,然后将搅拌混匀的鸡蛋液通过乙腈进行超声提取,最后进行磁性固相萃取净化,得到含氟虫腈及其代谢物的稀释溶液;通过乙腈进行超声提取和进行磁性固相萃取净化具体为:称取3g搅拌混匀的鸡蛋液加入50ml聚四氟乙烯离心管中,加入20ml乙腈,涡旋混匀1min,然后超声提取20min;加入1g氯化钠和3g无水硫酸镁,在转速为15000r/min离心10min,接着于-18℃放置20min,析出上清液;移取3ml上清液于100ml聚四氟乙烯离心管中,加入30ml水稀释,最后进行磁性固相萃取净化,磁性固相萃取净化时,磁性吸附剂用量为40mg,磁性吸附时间为15min,洗脱溶剂为乙腈,洗脱时间为15min,磁性固相萃取净化完成后,得到含氟虫腈及其代谢物的稀释溶液;b2、向步骤b1中所得的稀释溶液中加入30mg实施例1中制备的磁性纳米材料,采用涡旋混合仪涡旋混合5min,使稀释溶液中的氟虫腈及其代谢物被吸附到磁性纳米材料上;b3、用磁铁将吸附了氟虫腈及其代谢物的磁性纳米材料与溶液进行磁分离,弃去上清液,再用蒸馏水洗涤两次;再次进行磁分离,弃去上清液;b4、使用1ml乙腈将步骤b3中吸附了氟虫腈及其代谢物的磁性纳米材料上的氟虫腈及其代谢物的洗脱下来,然后进行磁分离,将磁性纳米材料分离出来,保留上清液;b5、吸取步骤b4中的上清液0.5ml于离心管中,加入0.5ml水,混匀后经0.22μm滤膜过滤,得待测样品;(3)采用超高效液相色谱-q-exactive高分辨质谱将步骤(2)中所得的待测样品进行检测分析。超高效液相色谱的条件为:dionexultimate3000超高效液相色谱仪配备wps-3000trs自动进样器、tcc-3000rs柱温箱和hpg-3400rs二元泵;色谱柱:accucoreaq(2.1×150mm,2.6μm粒径);流速:0.3ml/min;柱温:45℃;进样量:10μl;流动相由水(流动相a)和乙腈(流动相b)组成;梯度洗脱条件如下:0-3min,60%b;3-3.5min,60%b-90%b;3.5-4.5min,90%b;4.5-6min,90%b-60%b;6-9min,10%b;高分辨质谱的条件为:鞘气流量:35个任意单位;辅助气流量:15个任意单位;尾吹气流量:1个任意单位;喷雾电压:3.0kv(负离子模式);扫描范围:m/z为200-600;毛细管温度:350℃;s透镜rf:50个任意单位;辅助气温度:350℃;一级质谱全扫描分辨率:r=70000;c-trap最大容量(targetagc):3e6;c-trap最长等待时间:200ms;数据依赖二级全扫描(dd-ms2)分辨率:r=17500;c-trap最大容量(targetagc):5e4;c-trap最长等待时间:50ms;动态排除:10s。应用实施例2中的检测方法进行样品分析应用实施例2中的检测方法对实施例1中应用的20个鸡蛋样品中的氟虫腈及其代谢物进行分析,样品检测结果如下表5所示:表5样品检测结果氟虫腈(μg/kg)氟甲腈(μg/kg)氟虫腈亚砜(μg/kg)氟虫腈砜(μg/kg)1未检出未检出未检出未检出2未检出未检出未检出未检出3未检出未检出未检出未检出4未检出未检出未检出未检出5未检出未检出未检出未检出6未检出未检出未检出0.5937未检出未检出未检出未检出8未检出未检出未检出未检出9未检出未检出未检出未检出10未检出未检出未检出1.8111未检出未检出未检出未检出12未检出未检出未检出2.2113未检出未检出未检出未检出14未检出未检出未检出未检出15未检出未检出未检出未检出16未检出未检出未检出未检出17未检出未检出未检出未检出18未检出未检出未检出未检出19未检出未检出未检出未检出20未检出未检出未检出未检出由上表8可以看出,氟虫腈、氟甲腈和氟虫腈亚砜均未检测到,有3个样品(样品6、样品10和样品12)中检测出了氟虫腈砜,浓度分别为0.593μg/kg、1.81μg/kg和2.21μg/kg,与实施例1的检测结果基本相同,且低于欧盟规定的最高限量(0.005mg/kg),检测灵敏度和检测精确度均高于现有技术中的检测方法。实施例3:一种鸡蛋中氟虫腈及其代谢物的净化和检测方法,包括下述步骤:(1)制备磁性纳米材料a1、取0.72g柠檬酸钠、4.8g乙酸钠和3.0gfecl3·6h2o,用600ml乙二醇溶解后加入高压反应釜中,在200℃反应12h;将反应生成的fe3o4纳米颗粒在外加磁场的作用下磁分离,经蒸馏水洗涤2次,无水乙醇洗涤1次,-50℃冷冻干燥12h,得fe3o4纳米粒子;a2、称取步骤a1中制备的0.3g的fe3o4纳米粒子加入反应容器内,加入150ml盐酸(0.1m)超声15min;然后用蒸馏水洗涤4次;向反应容器内加入240ml甲醇、60ml水和3.0ml氨水溶液,然后在不断搅拌下逐滴加入3ml的teos,在室温下反应24h;在外加磁场的作用下磁分离,经无水乙醇和蒸馏水各洗涤3次;得fe3o4@nsio2纳米粒子;取少量fe3o4@nsio2纳米粒子于-50℃冷冻干燥12h,用于表征;a3、将0.3g步骤a2中合成的fe3o4@nsio2粒子中加入240ml水、180ml无水乙醇、1.8gctab和3.3ml氨水溶液超声分散15min,然后不断搅拌下逐滴加入3ml的teos,室温下反应24h;用盐酸-乙醇溶液(5:95,v/v)洗去残留的ctab,经无水乙醇和水各洗涤3次,-50℃冷冻干燥12h,得fe3o4@nsio2@msio2纳米粒子;a4、称取0.5g步骤a3中合成的fe3o4@nsio2@msio2磁性纳米粒子,将其分散在150ml的tris溶液(10mm,ph8.5)中,加入2.5g多巴胺,室温机械搅拌反应多少时间生成磁性纳米材料,将生成的磁性纳米材料进行磁分离后,经蒸馏水洗涤三次,-50℃冷冻干燥12h,即得到fe3o4@nsio2@msio2-pda磁性纳米材料。(2)样品前处理b1、新鲜鸡蛋去壳后搅拌混匀,然后将搅拌混匀的鸡蛋液通过乙腈进行超声提取,最后进行磁性固相萃取净化,得到含氟虫腈及其代谢物的稀释溶液;通过乙腈进行超声提取和进行磁性固相萃取净化具体为:称取1g搅拌混匀的鸡蛋液加入50ml聚四氟乙烯离心管中,加入5ml乙腈,涡旋混匀1min,然后超声提取10min;加入1g氯化钠和3g无水硫酸镁,在转速为12000r/min离心6min,接着于-18℃放置25min,析出上清液;移取5ml上清液于100ml聚四氟乙烯离心管中,加入30ml水稀释,最后进行磁性固相萃取净化,磁性固相萃取净化时,磁性吸附剂用量为5mg,磁性吸附时间为15min,洗脱溶剂为乙腈,洗脱时间为5min,磁性固相萃取净化完成后,得到含氟虫腈及其代谢物的稀释溶液;b2、向步骤b1中所得的稀释溶液中加入30mg实施例1中制备的磁性纳米材料,采用涡旋混合仪涡旋混合5min,使稀释溶液中的氟虫腈及其代谢物被吸附到磁性纳米材料上;b3、用磁铁将吸附了氟虫腈及其代谢物的磁性纳米材料与溶液进行磁分离,弃去上清液,再用蒸馏水洗涤两次;再次进行磁分离,弃去上清液;b4、使用1ml乙腈将步骤b3中吸附了氟虫腈及其代谢物的磁性纳米材料上的氟虫腈及其代谢物的洗脱下来,然后进行磁分离,将磁性纳米材料分离出来,保留上清液;b5、吸取步骤b4中的上清液0.5ml于离心管中,加入0.5ml水,混匀后经0.22μm滤膜过滤,得待测样品;(3)采用超高效液相色谱-q-exactive高分辨质谱将步骤(2)中所得的待测样品进行检测分析。超高效液相色谱的条件为:dionexultimate3000超高效液相色谱仪配备wps-3000trs自动进样器、tcc-3000rs柱温箱和hpg-3400rs二元泵;色谱柱:accucoreaq(2.1×150mm,2.6μm粒径);流速:0.3ml/min;柱温:30℃;进样量:2μl;流动相由水(流动相a)和乙腈(流动相b)组成;梯度洗脱条件如下:0-3min,60%b;3-3.5min,60%b-90%b;3.5-4.5min,90%b;4.5-6min,90%b-60%b;6-9min,10%b;高分辨质谱的条件为:鞘气流量:35个任意单位;辅助气流量:15个任意单位;尾吹气流量:1个任意单位;喷雾电压:1.50kv(负离子模式);扫描范围:m/z为200-600;毛细管温度:350℃;s透镜rf:50个任意单位;辅助气温度:350℃;一级质谱全扫描分辨率:r=70000;c-trap最大容量(targetagc):3e6;c-trap最长等待时间:200ms;数据依赖二级全扫描(dd-ms2)分辨率:r=17500;c-trap最大容量(targetagc):5e4;c-trap最长等待时间:50ms;动态排除:10s。应用实施例3中的检测方法进行样品分析应用实施例3中的检测方法对实施例1中应用的20个鸡蛋样品中的氟虫腈及其代谢物进行分析,样品检测结果如下表6所示:表6样品检测结果氟虫腈(μg/kg)氟甲腈(μg/kg)氟虫腈亚砜(μg/kg)氟虫腈砜(μg/kg)1未检出未检出未检出未检出2未检出未检出未检出未检出3未检出未检出未检出未检出4未检出未检出未检出未检出5未检出未检出未检出未检出6未检出未检出未检出0.5767未检出未检出未检出未检出8未检出未检出未检出未检出9未检出未检出未检出未检出10未检出未检出未检出1.9211未检出未检出未检出未检出12未检出未检出未检出2.0513未检出未检出未检出未检出14未检出未检出未检出未检出15未检出未检出未检出未检出16未检出未检出未检出未检出17未检出未检出未检出未检出18未检出未检出未检出未检出19未检出未检出未检出未检出20未检出未检出未检出未检出由上表12可以看出,氟虫腈、氟甲腈和氟虫腈亚砜均未检测到,有3个样品(样品6、样品10和样品12)中检测出了氟虫腈砜,浓度分别为0.576μg/kg、1.92μg/kg和2.05μg/kg,与实施例1和实施例2的检测结果基本相同,且低于欧盟规定的最高限量(0.005mg/kg),检测灵敏度和检测精确度均高于现有技术中的检测方法。以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本
技术领域:
的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1