一种血管生成抑肽的检测方法与流程
本发明涉及多肽检测领域,尤其涉及一种血管生成抑肽的检测方法。
背景技术:
血管生成抑肽(fpat)是一种与整合素有亲和能力的或结合能力的高效血管生成抑制剂,将抑制血管生成的内皮抑素的氨基酸序列c端加上含有精氨酸-甘氨酸-天冬氨酸的多肽序列。
fpat是利用化学合成方法合成的一段具有十五个氨基酸的多肽片段,具体结构为asp-ser-gly-glu-gly-asp-phe-leu-ala-glu-gly-gly-gly-val-arg·xc2h4o2,分子式为c60h92n18o25·xc2h4o2。在化学合成的过程中,往往会引入很多工艺杂质,如异构体杂质、缺失肽、插入肽、断裂肽、消旋肽、去酰胺肽、氨基酸侧链的不完全脱保护所形成的副产物、氧化肽、二硫键交换产物等。由于fpat在抗肿瘤治疗方面有着较大的潜力,特别是血液有关的恶性肿瘤,包括急性淋巴母细胞白血病、慢性淋巴细胞白血病、急性骨髓性粒细胞白血病、慢性骨髓性粒细胞白血病、急性恶性骨髓硬化症、多发性骨髓瘤、真性红细胞增多症、瓦尔登斯特伦巨球蛋白血症、何杰金淋巴瘤、非何杰金淋巴瘤等,因此对fpat的含量进行高效准确的测定尤为重要。
目前为止,尚未建立一套全面、有效、快速、稳定分析血管生成抑肽的检测方法。
技术实现要素:
针对现有技术存在的问题,本发明提供一种血管生成抑肽的检测方法,实现对血管生成抑肽的快速准确检测。
本发明采用以下技术方案:
本发明提供一种血管生成抑肽的检测方法,利用超高效液相色谱分析方法对血管生成抑肽样品进行分析,其中,所述超高效液相色谱分析的条件包括:以庚烷磺酸钠-乙腈为流动相a,以乙腈为流动相b进行梯度洗脱。
本发明经过大量实验发现,利用超高效液相色谱分析方法进行分析时,以庚烷磺酸钠-乙腈为流动相a,以乙腈为流动相b进行梯度洗脱,血管生成抑肽主峰与杂质峰的分离效果较好,比常用的三氟乙酸等流动相体系分离效果好。
进一步地,所述流动相a中庚烷磺酸钠与乙腈的体积比为85:15,其中庚烷磺酸钠用磷酸调ph值至2.50。
进一步地,所述梯度洗脱的条件为:0-8min,95%流动相a,5%流动相b;8-8.1min,80%流动相a,20%流动相b;8.1-12min,95%流动相a,5%流动相b。
在上述优选条件下,可以使主峰与前后杂质之间的分离度、理论塔板数和拖尾因子均满足要求,实现了快速准确的检测。
进一步地,所述梯度洗脱过程中,流动相的流速为0.35±0.02ml/min。
进一步地,色谱柱以十八烷基硅烷键合硅胶为填充剂(即采用c18柱),柱温为60±5℃,优选为60℃。
进一步地,所述色谱柱的规格为2.1×100mm,1.7μm,
进一步地,进样浓度1.0±0.2mg/ml,进样量0.8±0.2ul,检测波长为210±5nm。
进一步地,所述血管生成抑肽样品以供试品溶液的形式提供,所述供试品溶液为血管生成抑肽的水溶液。
在本发明的优选实施方式中,所述血管生成抑肽的检测方法具体包括以下步骤:
将血管生成抑肽样品加水溶解配制成样品溶液;
利用超高效液相色谱对所述样品溶液进行检测分析,其中,色谱条件如下:
色谱柱为c18柱;
流动相a为庚烷磺酸钠与乙腈体积比为85:15的混合溶液,其中庚烷磺酸钠用磷酸调ph值至2.50,流动相b为乙腈;
梯度洗脱的条件为:0-8min,95%流动相a,5%流动相b;8-8.1min,80%流动相a,20%流动相b;8.1-12min,95%流动相a,5%流动相b;
流动相流速为0.35ml/min,柱温为60℃,进样浓度1.0mg/ml,进样量0.8ul,检测波长为210nm。
本发明所述检测方法也适用于血管生成抑肽原料药中杂质的检测,所述杂质包括下列物质中的一种或多种:
lgx3:asp-ser-glu-gly-asp-phe-leu-ala-glu-gly-gly-gly-val-arg。
本发明还提供上述血管生成抑肽的检测方法在血管生成抑肽的生产和质量控制中的应用。
本发明提供了一种血管生成抑肽的检测方法,可实现对血管生成抑肽的快速准确检测,相对于传统的高效液相色谱分析方法可节省75%的检测时间,而且该方法主峰与前后杂质之间的分离度、理论塔板数和拖尾因子均可以满足方法要求。
附图说明
图1为本发明实施例1中系统适用性溶液的超高效液相色谱分析谱图;
图2为本发明实施例2中fpat的超高效液相色谱分析谱图;
图3为本发明实施例3中系统适用性溶液的超高效液相色谱分析谱图;
图4为本发明实施例4中系统适用性溶液的超高效液相色谱分析谱图;
图5为本发明实施例5中系统适用性溶液在柱温60℃下的超高效液相色谱分析谱图;
图6为本发明实施例5中系统适用性溶液在柱温35℃下的超高效液相色谱分析谱图;
图7为本发明实施例5中系统适用性溶液在柱温45℃下的超高效液相色谱分析谱图;
图8为本发明实施例6中系统适用性溶液的超高效液相色谱分析谱图;
图9为本发明实施例7中系统适用性溶液的超高效液相色谱分析谱图;
图10为本发明实施例7中fpat加速6月样品的超高效液相色谱分析谱图;
图11为本发明实施例8中系统适用性溶液的超高效液相色谱分析谱图;
图12为本发明对比例1中fpat的超高效液相色谱分析谱图;
图13为本发明对比例2中fpat的超高效液相色谱分析谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
若未特别指明,本发明实施例中所用的实验试剂和材料等均可市售获得。
若未具体指明,本发明实施例中所用的技术手段均为本领域技术人员所熟知的常规手段。
实施例1
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,色谱柱以十八烷基硅烷键合硅胶为填充剂(2.1×100mm,1.7μm,
表1洗脱梯度
系统适用性溶液制备:取lgx3,用水进行溶解,制成浓度为0.1mg/ml的lgx3溶液;取fpat原料,用lgx3溶液进行溶解,制成1mg/ml的系统适用性溶液。
进样检测:将待检测溶液注入超高效液相色谱仪,进行梯度洗脱,记录色谱图,结果如图1所示,从图中计算出原料主峰与后杂lgx3峰分离度1.846,与前杂分离度1.669,主峰塔板数52870。
实施例2
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,c18色谱柱(2.1×100mm,1.7μm,
表2洗脱梯度
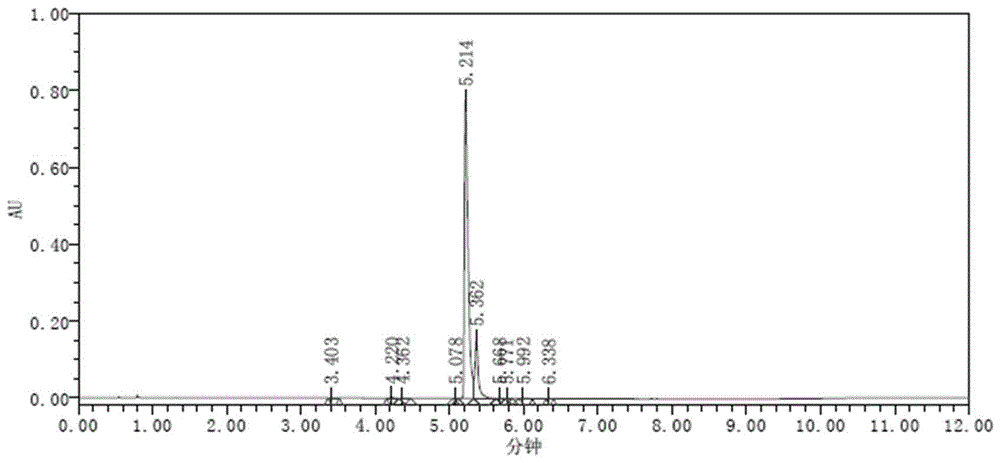
将fpat原料溶于水制成1mg/ml的样品溶液进行进样检测,结果如图2所示,从图中看出主峰很快析出,没有挂柱。
实施例3
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,c18色谱柱(2.1×100mm,1.7μm,
表3洗脱梯度
对系统适用性溶液进行测定,结果如图3所示,理论板数按fpat峰计算为146510;紧邻主峰后杂lgx3与fpat峰的分离度1.60。但原料主峰包含杂质,拖尾严重。
实施例4
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,其检测仪器与色谱条件与实施例3的区别在于以0.01mol/l庚烷磺酸钠(用磷酸调ph值至3.50)为流动相a,其余相同。
对系统适用性溶液进行测定,结果如图4所示,从图中看出原料主峰峰型不好,包有杂质峰。
实施例5
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,c18色谱柱(2.1×100mm,1.7μm,
对系统适用性溶液进行测定,结果如图5-7所示,从图中看出其他条件不变,60℃时杂质检出最多。
实施例6
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,c18色谱柱(2.1×100mm,1.7μm,
表4洗脱梯度
对系统适用性溶液进行测定,结果如图8所示,从图中看出,流速0.2ml/min洗脱时,lgx3峰与fpat峰的分离度没有流速0.35ml/min时对后杂的分离效果好。
实施例7
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,c18色谱柱(2.1×100mm,1.7μm,
对系统适用性溶液、fpat加速6月样品进行测定,结果如图9-10所示,结果为:系统适用性溶液,理论板数按fpat峰计算为18981.60,lgx3峰与fpat峰的分离度1.952,但主峰拖尾严重,usp拖尾为4.058;fpat加速6月样品,fpat峰与后杂分离度5.478,未分离出lgx3杂质,主峰拖尾严重,usp拖尾为4.290。
实施例8
本实施例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,c18色谱柱(2.1×100mm,1.7μm,
表5洗脱梯度
对系统适用性溶液进行测定,结果如图11所示,从图中看出原料主峰与lgx3峰无法分离。
对比例1
本对比例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,色谱柱以十八烷基硅烷键合硅胶为填充剂(2.1×100mm,1.7μm,
表6洗脱梯度
对fpat原料进行测定,结果如图12所示,从图中看出主峰峰型不好,包有杂质峰。
对比例2
本对比例提供一种血管生成抑肽fpat的超高效液相色谱分析方法,具体如下:
检测仪器与色谱条件:
watersh-class超高效液相色谱仪,色谱柱以十八烷基硅烷键合硅胶为填充剂(2.1×100mm,1.7μm,
表7洗脱梯度
对fpat原料进行测定,结果如图13所示,从图中看出主峰峰型不好,包有杂质峰。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!