一种植物叶片中多环芳烃含量的分析方法
1.本发明涉及物质和检测技术领域,具体涉及一种植物叶片中多环芳烃含量的分析方法。
背景技术:
2.多环芳烃(polycyclic aromatic hydrocarbons,pahs)是一类大分子非极性有机物,具有致癌、致畸、致突变的特性。因此,部分国家环保局已将其中16种无分支结构的pahs列为优先控制污染物。环境中的pahs主要由有机物高温不完全燃烧产生,其通过大气沉降作用沉积在植物叶片上。植物叶片中pahs的含量水平,可评价某地区植物受pahs污染的程度,亦可间接评价空气中pahs的污染程度。所以,准确检测植物样品pahs的含量显得极为重要。pahs在植物叶片中含量极低,且叶片中存在大量的色素、植物碱及脂类物质等,这些物质均会干扰叶片pahs的分析,给pahs分析带来困难。
3.植物叶片中pahs含量分析主要包括:样品萃取、活化和检测三个主要步骤。目前,样品萃取的方法主要有:索氏萃取,加速溶剂萃取,超声萃取,微波辅助萃取等方法。其中:加速溶剂萃取法具有使用溶剂量少,节省时间,操作简单等优点,但在萃取溶剂的选择方面,传统的方法一般采用丙酮:二氯甲烷、甲苯、正己烷等,这些溶剂在土壤pahs萃取使用广泛,且萃取效果好,但在植物样品中却不适用,易将植物样品中的色素、脂类等物质共萃取出来,干扰目标物质的分析。而其他萃取方法如索氏萃取,具有萃取时间长,操作繁琐,使用溶剂量大等缺点,不符合当今低碳环保的主题。同时,现有技术中,一般采用硅胶或氧化铝填充柱活化萃取液,使用填充柱活化,不仅填充柱子费时费力,而且数据重复性不佳。检测方法方面,现有技术主要采用气相色谱
‑
质谱或液相色谱检测,其检测灵敏度不高。传统的测定方法,如索氏萃取+皂化+硅胶填充柱活化+检测分析,其测定一个样品花费的时间至少需5个小时以上,且操作繁琐。
技术实现要素:
4.本发明提出一种植物叶片中多环芳烃含量的分析方法,以解决现有技术中存在的一个或多个技术问题,至少提供一种有益的选择或创造条件。
5.为克服上述技术问题,本发明采用的技术方案如下:
6.一种植物叶片中多环芳烃含量的分析方法,包括以下步骤:
7.s1.采用加速溶剂萃取法对植物叶片粉末进行萃取,得萃取液,所述萃取采用的溶剂为乙腈;
8.s2.对所述萃取液进行活化处理,得活化液;
9.s3.测定所述活化液中的多环芳烃含量。
10.本发明采用加速溶剂萃取法对植物叶片粉末进行萃取,所采用的萃取溶剂为乙腈,与传统方法采用的溶剂不同,传统方法一般采用甲苯、丙酮、正己烷等,这些溶剂不仅毒性大,危害操作人员的身体健康,且杂质共萃取率高,干扰pahs分析,不符合pahs含量分析
的要求。乙腈为中极性的有机溶剂,不仅可有效地提取植物叶片中的pahs,且杂质的共萃取率较低,共萃取的杂质的量不会对pahs分析造成干扰。
11.进一步地,所述多环芳烃为萘、苊、二氢苊、芴、菲、蒽、荧蒽、芘、苯[a]并蒽、屈、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、印并[1,2,3
‑
cd]芘、二苯并[a,h]蒽和苯并[g h i]苝。该16种无分支结构的pahs为环保部门优先控制的污染物。
[0012]
作为上述方案的进一步改进,步骤s1中所述植物叶片粉末的制备方法,包括以下步骤:
[0013]
将采集的植物叶片先用自来水洗净,再用去离子水冲洗三遍,得活化植物叶片;将所述活化植物叶片置于90
‑
110℃烘箱中烘烤3
‑
5小时后,进行粉碎,得植物叶片粉末,所述植物叶片粉末的粒径为过30
‑
60目筛;将所述植物叶片粉末避光保存待用。具体地,对植物叶片进行反复冲洗,可保证将附着于叶片表面的泥土及灰尘彻底清除。过筛后的植物叶片粉末置于棕色玻璃瓶进行避光保存,存放温度为
‑
20℃。
[0014]
作为上述方案的进一步改进,步骤s1中所述加速溶剂萃取法对植物叶片粉末进行萃取的方法为:
[0015]
取1g所述植物叶片粉末和1g经硝酸激活的铜粉混合后加入到34ml萃取池中,并用硅藻土填满萃取池;在萃取池中加入10μl浓度为10μg/ml的间
‑
三联苯作为内标、溶剂乙腈;在萃取温度为120℃,萃取压力为10mpa的条件下,进行静态萃取2次,每次萃取时间为5min;用20.4ml溶剂乙腈对萃取池进行冲洗,并用氮气吹扫90s,得萃取液。
[0016]
具体地,本发明中所得的萃取液含干扰杂质少,可省略皂化的步骤,减少劳力和时间的花费,降低pahs损失。传统的方法一般要用氢氧化钾或浓硫酸皂化,皂化过程中不仅很容易造成pahs的损失,且所用试剂属于强酸、强碱,操作过程存在一定的危险性。
[0017]
进一步地,所述硝酸为浓度大于8mol/l的浓硝酸。
[0018]
作为上述方案的进一步改进,步骤s2中所述活化处理采用c18固相萃取小柱对所述萃取液进行,包括以下步骤:
[0019]
将所述萃取液在30
‑
40℃水浴中旋转蒸发,得蒸干萃取液,优选地,将所述萃取液转移至旋转蒸发瓶中旋转蒸发;
[0020]
先后用5ml环己烷、5ml甲醇和15ml去离子水以2ml/min的流速淋洗c18固相萃取小柱,得活化c18固相萃取小柱;
[0021]
将所述蒸干萃取液用10ml质量浓度为30%的丙酮转移至所述活化c18固相萃取小柱,并以2ml/min的速度重复过柱三次。
[0022]
具体地,本发明采用质量浓度为30%的丙酮作为pahs的转移和淋洗溶剂,可较完全的把pahs从旋转蒸发瓶中转移至c18固相萃取小柱上,并将杂质洗脱去除。相对传统方法采用两种溶剂,一种将pahs从旋转蒸发瓶转移至c18小柱,一种用于杂质洗脱,本发明只用一种溶剂进行萃取液的转移和淋洗,简化了实验操作。同时,采用c18固相萃取小柱活化萃取液,具有良好的活化效果。
[0023]
作为上述方案的进一步改进,所述c18固相萃取小柱出现堵塞时,将其套在离心管内,以3000
‑
4000rpm离心1
‑
3min进行疏通。具体地,对于杂质较高的植物叶片,c18固相萃取小柱易于堵塞,当所述c18固相萃取小柱出现堵塞时,优选的疏通方法为:将c18固相萃取小柱套在15ml玻璃离心管离里以3500rpm离心2min进行疏通;若小柱持续堵塞,可直接
3500rpm离心进行洗脱。
[0024]
作为上述方案的进一步改进,步骤s3中所述活化液在进行测定前还包括依次进行的洗脱、去水和定容的步骤。
[0025]
进一步的,所述洗脱的步骤为:采用8ml环己烷对所述c18固相萃取小柱进行洗脱。
[0026]
进一步的,所述去水和定容的步骤为:采用无水硫酸钠进行去水,并用氮气进行吹扫,定容至1ml。
[0027]
作为上述方案的进一步改进,步骤s3中所述测定活化液中的多环芳烃含量采用气相色谱
‑
质谱连用的方法进行,其测定条件参数为:
[0028]
测定温度:进样口温度为280℃,检测器温度为300℃,离子源温度为300℃,四级杆温度为180℃;
[0029]
升温制度:60℃保持1min,以10℃/min升温至120℃,再以4℃/min升至300℃,保持10min;
[0030]
载气:高纯氦气,柱压10psi恒压;
[0031]
扫描模式:sim,扫描范围45
‑
550aum;
[0032]
进样方式:不分流,进样量:1μl;
[0033]
数据采集和处理:chemistation enhance化学工作站
[0034]
多环芳烃定量方法:外表单点定量。
[0035]
具体地,在特定的测定参数与模式下,采用气相色谱
‑
质谱连用的方法测定所述活化液中的多环芳烃,不仅可提高检测准确度,更可实现对难以准确测量的同分异构体进行定性和定量测定,同时,还可提高检测效率。同时,采用气相色谱
‑
质谱连用的方法测定所述活化液中的多环芳烃,具有良好的检测效果,检测灵敏度高。可运用保留时间和全扫描质谱定性,选择离子扫描模式定量,实现准确的对同分异构体(如苯并[b]荧蒽和苯并[k]荧蒽)进行定性和定量分析。
[0036]
本技术相对于现有技术,至少具有如下技术效果或优点:
[0037]
本发明在对植物叶片粉末进行萃取时,采用乙腈作为萃取溶剂,该溶剂与传统方法溶剂相比,既可保证pahs的回收率高,又能减少杂质的共萃取,同时不干扰pahs的分析。制得的萃取液含干扰杂质少,并可省略皂化的步骤,减少劳力和时间的花费,降低pahs损失。同时,萃取过程中使用有机溶剂少,节约成本的同时可降低环境污染。
[0038]
本发明操作简单、省时、分析速度快,平均每个样品萃取、活化和测定的时间仅需2小时左右,大大提高了测定分析的效率,且检测灵敏度高,适用于样品量小于1g的植物叶片检测。
附图说明
[0039]
图1是pahs的色谱图。
[0040]
图中:各色谱峰分别代表,1、萘;2、苊;3、二氢苊;4、芴;5、菲;6、蒽;7、荧蒽;8、芘;9、苯[a]并蒽;10、屈;11、苯并[b]荧蒽;12、苯并[k]荧蒽;13、苯并[a]芘;14、印并[1,2,3
‑
cd]芘;15、二苯并[a,h]蒽;16、苯并[g hi]苝。
具体实施方式
[0041]
以下通过实施例对本发明进行具体描述,以便于所属技术领域的人员对本发明的理解,有必要在此特别指出的是,实施例只是用于对本发明做进一步说明,不能理解为对本发明保护范围的限制,所属领域技术人员,根据上述发明内容对本发明作出的非本质性的改进和调整,应仍属于本发明的保护范围,同时,下述所提及的原料未详细说明的,均为市售产品,未详细提及的工艺步骤或制备方法均为本领域技术人员所知晓的工艺步骤或制备方法。
[0042]
实施例1
[0043]
本实施例为植物叶片的加标回收实验,以测定植物叶片中多环芳烃的回收率及操作的可重复性,本实施例的植物叶片采自深圳湾红树林的桐花树叶片。
[0044]
一种植物叶片中多环芳烃含量的分析方法,包括以下步骤:
[0045]
(1)植物叶片的预处理:将200g新鲜的植物叶片置于烘箱中100℃连续4小时烘干,烘干的植物叶片用粉碎机粉碎,过40目筛,得植物叶片粉末,过筛后的植物叶片粉末置于棕色玻璃瓶中保存待用。
[0046]
(2)多环芳烃的萃取:采用美国戴安加速溶剂萃取仪ase
‑
100对所述植物叶片粉末进行萃取,萃取步骤为:先称取1g植物叶片粉末和1g经10mol/l的浓硝酸激活的铜粉,将二者在玻璃质研钵中混合均匀,再加入适量的硅藻土(分析纯,使用前550℃烘4h)再混合均匀。然后将其加入到加速溶剂萃取仪的萃取池(34ml)中,加入质量浓度为10μl1000μg/ml的间
‑
三联苯和50μl质量浓度为200μg/ml的16种pahs混标(合10μg间
‑
三联苯,10μg pahs混标),再用硅藻土填满萃取池。设定萃取条件参数:溶剂是乙腈,萃取温度为120℃,萃取压力10mpa,静态萃取时间5min循环2次,冲洗体积为萃取池体积60%,氮气吹扫时间90s,制得萃取液。
[0047]
(3)c18固相萃取小柱活化萃取液:采用waters c18固相萃取小柱(3ml,500mg)进行活化,活化步骤为:
[0048]
旋转蒸发:将萃取液转移至旋转蒸发瓶中采用35℃水浴进行旋转蒸发,得蒸干萃取液,待用。
[0049]
c18固相萃取小柱预处理:先后用5ml环己烷、5ml甲醇和15ml去离子水以2ml/min流速淋洗柱子。
[0050]
上样及预淋洗:将蒸干萃取液用10ml质量浓度为30%丙酮(先加3ml丙酮将旋转蒸发瓶冲洗干净,再加7ml去离子水)转移至c18固相萃取小柱,并以2ml/min的速度过柱,此步骤重复三次。若过柱过程柱子出现堵塞,则可将柱子套在小试管内,以3500rpm离心1min。
[0051]
(4)洗脱:用8ml环己烷对c18固相萃取小柱进行洗脱,将目标化合物从c18固相萃取小柱上洗脱下来。
[0052]
(5)去水:洗脱下来的溶液中含有少量的水,加入适量无水硫酸钠去水。
[0053]
(6)定容:将已去水的洗脱液用氮气吹扫,定容至1ml备用。
[0054]
(7)气相色谱
‑
质谱连用测定待测样品中的多环芳烃:测定条件参数:色谱柱,hp
‑
5ms(30m长
×
0.25mm直径.
×
0.25μm膜厚);进样口温度:280℃,检测器温度:300℃,离子源温度:300℃,四级杆温度:180℃;升温程序:60℃保持1min,以10℃/min升至120℃,再以4℃/min升至300℃,保持10min;载气:高纯氦气,柱压10psi恒压;扫描模式:sim,扫描范围
45
‑
550aum;进样量:不分流1μl;数据采集和处理:chemistation enhance化学工作站;pahs定量方法:标准曲线法。
[0055]
本实施例所测的植物叶片中的pahs成分如图1所示,图1中横坐标为时间,纵坐标为电压,由图1的色谱图可知,共测出16种pahs,具体为萘、苊、二氢苊、芴、菲、蒽、荧蒽、芘、苯[a]并蒽、屈、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、印并[1,2,3
‑
cd]芘、二苯并[a,h]蒽和苯并[g h i]苝。
[0056]
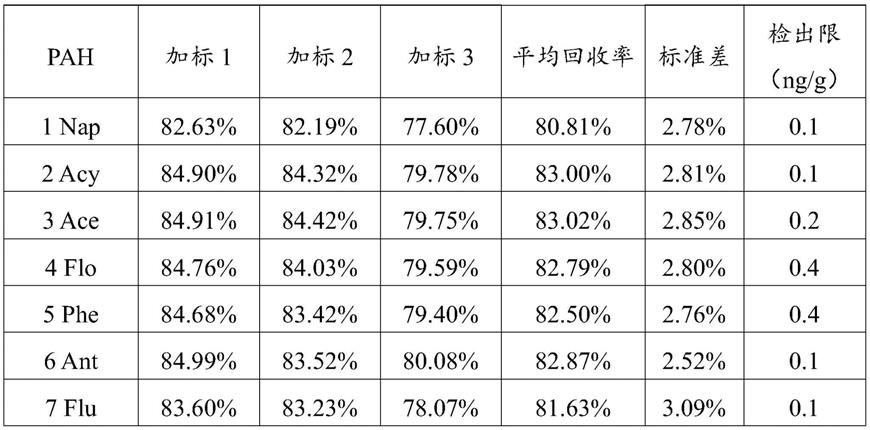
本实施例所测的植物叶片中的各pahs的回收率如表1所示。
[0057]
表1植物叶片中pahs的回收率表
[0058][0059][0060]
表1中:1nap:萘;2acy:苊;3ace:二氢苊;4flo:芴;5phe:菲;6ant:蒽;7flu:荧蒽;8pyr:芘;9baa:苯并[a]蒽;10chr:屈;11bbf:苯并[b]荧蒽12bkf:苯并[k]荧蒽;13bap:苯并[a]芘;14i1p:印并[1,2,3
‑
cd]芘;15daa:二苯并[a,h]蒽;16bgp:苯并[g h i]苝。
[0061]
其中:加标1、加标2和加标3分别代表三次加标回收实验测定值。
[0062]
从表1中各pahs的回收率结果可知,16种pahs及内标间三联苯的回收率在80
‑
117%之间,且标准差较小,符合痕量有机物定量分析的质量控制标准。
[0063]
实施例2
[0064]
本实施例选取深圳坝光红树植物叶片,对该植物叶中的pahs含量进行测定分析。
[0065]
一种植物叶片中多环芳烃含量的分析方法,包括以下步骤:
[0066]
(1)植物叶片的预处理:将200g新鲜的植物叶片置于烘箱中100℃连续4小时烘干,烘干的植物叶片用粉碎机粉碎,过40目筛,得植物叶片粉末,过筛后的植物叶片粉末置于棕色玻璃瓶中保存待用。
[0067]
(2)多环芳烃的萃取:采用美国戴安加速溶剂萃取仪ase
‑
100对所述植物叶片粉末进行萃取,萃取步骤为:先称取1g植物叶片粉末和1g经10mol/l的浓硝酸激活的铜粉,将二者在玻璃质研钵中混合均匀,再加入适量的硅藻土(分析纯,使用前550℃烘4h)再混合均匀。然后将其加入到加速溶剂萃取仪的萃取池(34ml)中,加入质量浓度为10μl1000μg/ml的间
‑
三联苯(合10μg间
‑
三联苯),再用硅藻土填满萃取池。设定萃取条件参数:溶剂是乙腈,萃取温度为120℃,萃取压力10mpa,静态萃取时间5min循环2次,冲洗体积为萃取池体积60%,氮气吹扫时间90s,制得萃取液。
[0068]
(3)c18固相萃取小柱活化萃取液:采用waters c18固相萃取小柱(3ml,500mg)进行活化,活化步骤为:
[0069]
旋转蒸发:将萃取液转移至旋转蒸发瓶中采用35℃水浴进行旋转蒸发,得蒸干萃取液,待用。
[0070]
c18固相萃取小柱预处理:先后用5ml环己烷、5ml甲醇和15ml去离子水以2ml/min流速淋洗柱子。
[0071]
上样及预淋洗:将蒸干萃取液用10ml质量浓度为30%丙酮(先加3ml丙酮将旋转蒸发瓶冲洗干净,再加7ml去离子水)转移至c18固相萃取小柱,并以2ml/min的速度过柱,此步骤重复三次。若过柱过程柱子出现堵塞,则可将柱子套在小试管内,以3500rpm离心1min。
[0072]
(4)洗脱:用8ml环己烷对c18固相萃取小柱进行洗脱,将目标化合物从c18固相萃取小柱上洗脱下来。
[0073]
(5)去水:洗脱下来的溶液中含有少量的水,加入适量无水硫酸钠去水。
[0074]
(6)定容:将已去水的洗脱液用氮气吹扫,定容至1ml备用。
[0075]
(7)气相色谱
‑
质谱连用测定待测样品中的多环芳烃:测定条件参数:色谱柱,hp
‑
5ms(30m长
×
0.25mm直径.
×
0.25μm膜厚);进样口温度:280℃,检测器温度:300℃,离子源温度:300℃,四级杆温度:180℃;升温程序:60℃保持1min,以10℃/min升至120℃,再以4℃/min升至300℃,保持10min;载气:高纯氦气,柱压10psi恒压;扫描模式:sim,扫描范围45
‑
550aum;进样量:不分流1μl;数据采集和处理:chemistation enhance化学工作站;pahs定量方法:标准曲线法。
[0076]
本实施例所测得的内标间
‑
三联苯的回收率在48%
‑
105%之间,植物叶片中pahs含量如表2,由表2可知,同批样植物叶片的回收率重复性较佳。
[0077]
表2红树植物叶片中的pahs含量表
[0078][0079]
表2中:1nap:萘;2acy:苊;3ace:二氢苊;4flo:芴;5phe:菲;6ant:蒽;7flu:荧蒽;8pyr:芘;9baa:苯并[a]蒽;10chr:屈;11bbf:苯并[b]荧蒽12bkf:苯并[k]荧蒽;13bap:苯并[a]芘;14i1p:印并[1,2,3
‑
cd]芘;15daa:二苯并[a,h]蒽;16bgp:苯并[g h i]苝。
[0080]
其中:a、b、c分别代表同一植物叶片的三个不同取样点。
[0081]
本发明的植物叶片中多环芳烃含量的分析方法,采用乙腈作为萃取溶剂,丙酮进行洗脱,相对传统分析方法采用正己烷作为萃取溶剂,正己烷和二氯甲烷进行洗脱,本发明在保证pah回收率的前提下,所采用试剂毒性小,且用量少,更为节约与环保。
[0082]
显然,上述实施例仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1