一种测定血浆中扶正化瘀制剂五种有效成分浓度的方法与流程
1.本发明涉及生物样品色谱检测技术领域,尤其涉及一种采用uhplc-ms/ms法测定血液中扶正化瘀制剂五种有效成分浓度的方法。
背景技术:
2.慢性肝病病程长,病情复杂,但从中医上来说主要是机体感染疫毒,引起正邪相搏,如正气虚则不能祛邪,至病情缠绵久致顽疾,其基本病机为正虚邪实、虚实共存。正虚主要是气阴两虚,而邪实则为蕴热化湿,血瘀络阻,气阴两虚、湿热疫毒内留。从西医角度来说肝纤维化是指肝细胞发生坏死及炎症刺激时,肝脏内纤维结缔组织异常增生的病理过程。慢性肝病绝大多数都有肝纤维化,肝纤维化是慢性肝病最重要的病理特征,肝纤维化与肝硬化既有联系又有区别,从病理上看,仅有弥漫性肝纤维沉积增加称肝纤维化,而弥漫性肝纤维化同时伴有肝小叶结构被破坏则为肝硬化。从发病学看,肝纤维化是肝硬化的前期病变、是可逆的、而肝硬化是肝纤维化进一步发展的结果。从临床观察看,肝纤维化和肝硬化是连续的发展过程,肝纤维化一般不致肝功能障碍,但可有门静脉高压。肝纤维化是肝损伤后的修复过程,引起肝脏细胞外基质(ecm)和量的改变。
3.肝纤维化是各种慢性肝病发展为肝硬化所必经的病理过程,寻找阻止、延缓甚至逆转肝纤维化的药物,是肝病研究急待解决的问题。长期以来,中医药在抗肝纤维化方面发挥着重要的作用。中医认为,肝纤维化既有瘀滞的一面,也有正气虚损的一面,扶正化瘀是重要的治疗原则。扶正化瘀胶囊(片剂)正是针对慢性肝炎肝纤维化、肝硬化的“瘀血内结、正气虚弱”,在反复药理研究基础上而配伍组方研制的,由活血化瘀和益精补虚类药物组成。其中丹参活血化瘀,为君药;虫草菌丝补虚益精,桃仁去瘀破癓,为臣药;松花粉养肝散肝,绞股蓝清热解毒,为佐药;五味子酸温补肝,为使药。药理实验表明,该复方制剂具有抗肝纤维化,降低门脉压力,保护肝细胞,减轻脂质过氧化损伤,调整免疫功能,减轻肝脏炎症等功效。扶正化瘀制剂经临床验证可使肝纤维化分期运转率达52%~58.3%,因此,扶正化瘀制剂治疗肝硬化失代偿疗效明显。
4.扶正化瘀制剂是由丹参、发酵虫草菌粉、桃仁、松花粉、绞股蓝、五味子六味药组成,具有治疗肝、肺、肾纤维化的作用。目前上市有两种剂型:(1)扶正化瘀胶囊:2002年获得中国国家新药证书(国药证字z20020053),并批准国家药品标准ws3-459(z-79)-2005(z);(2)扶正化瘀片剂:2005年获得国家新药证书(国药证字z20050564),并批准国家药品标准ybz19332005-2009z。扶正化瘀制剂主要用于乙型肝炎肝纤维化属瘀血阻络,肝肾不足证者,症见胁下痞块,胁肋疼痛,面色晦暗,或见赤缕红斑,腰膝酸软,疲倦乏力,头晕目涩,舌质暗红或有瘀斑,苔薄或微黄,脉弦细。
5.整个扶正化瘀复方药效物质基础主要包含脂溶性的丹参酮类和水溶性的丹酚酸类、多糖、甾醇、多种氨基酸、多种维生素、挥发油、木脂素等。检测比格犬(beagle犬)血浆中扶正化瘀片的毒代动力学特征,评估药物主要成分在比格犬体内的暴露水平,对于帮助了解药物在人体内的作用效果具有重要意义。因此,本领域技术人员持续致力于研究测定动
物血浆中扶正化瘀制剂主要有效成分浓度的uhplc-ms/ms方法,其中该有效成分为以下中的一种或多种:五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素。
技术实现要素:
6.基于此,本发明所要解决的技术问题是提供了一种测定血液中扶正化瘀制剂有效成分浓度的方法,该方法包括以下步骤:
7.(1)待测样本预处理,获取待测液;
8.(2)有效成分标准曲线制备;以及
9.(3)液相色谱-质谱联用检测该待测液中该有效成分浓度,
10.其中,该液相色谱-质谱联用的色谱条件为:色谱柱:acquity uplc hss t3(2.1mm
×
100mm,1.8μm),流动相:水相(a)为酸水溶液,有机相(b)为甲醇或乙腈;洗脱方式:采用以下梯度程序洗脱:0-12min,5%b;12-14min,95%b;14-16min,5%b;
11.其中,该液相色谱-质谱联用的质谱条件为:电喷雾离子源(esi),采用正负离子模式检测,质量扫描范围m/z 100-800;
12.其中,该有效成分为以下中的一种或多种:五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素。
13.进一步地,该待测样本为哺乳动物的血浆。
14.进一步地,该哺乳动物为大鼠、小鼠或犬。
15.进一步地,该预处理包括以下步骤:
16.(1)吸取该待测样本,添加至第一96孔板中;
17.(2)在该第一96孔板中添加内标工作液;
18.(3)封板后涡旋混匀并离心,得到第一上清液;
19.(4)吸取该第一上清液,添加至第二96孔板中;
20.(5)在该第二96孔板中添加第一溶剂;以及
21.(6)封板后涡旋混匀并离心,得到该待测液。
22.进一步地,该有效成分标准曲线制备包括以下步骤:
23.(1)分别吸取校正标示样品和质控样品,分别添加至第一96孔板中;
24.(2)在该第一96孔板中分别添加内标工作液;
25.(3)封板后涡旋混匀并离心,得到第二上清液和第三上清液;
26.(4)分别吸取该第二上清液和第三上清液,分别添加至第二96孔板中;
27.(5)在该第二96孔板中分别添加第一溶剂;
28.(6)封板后涡旋混匀并离心,得到第四上清液和第五上清液;
29.(7)对该第四上清液和该第五上清液进行该液相色谱-质谱联用分析,得到数据;以及
30.(8)使用内标法对该数据进行处理,分别以该有效成分的浓度为横坐标,该有效成分和柳胺酚的峰面积比值为纵坐标,以1/x2为权重系数,利用加权最小二乘法进行线性回归,得到该有效成分的标准曲线。
31.进一步地,该校正标示样品的制备包括以下步骤:
32.(1)称量适量的有效成分,用第二溶剂溶解得到1mg/ml的有效成分储备液,将该有
效成分储备液用第二溶剂稀释成梯度浓度400~4*105ng/ml的校正标示样品工作液;以及
33.(2)将该校正标示样品工作液用空白基质稀释成梯度浓度为20~2*104ng/ml的该校正标示样品,该空白基质为不含该有效成分的该哺乳动物的血浆;
34.进一步地,该质控样品的制备包括以下步骤:
35.(1)称量适量的有效成分,用第二溶剂溶解得到1mg/ml的有效成分储备液,将该有效成分储备液用第二溶剂稀释成梯度浓度400~3*105ng/ml的质控工作液;以及
36.(2)将该质控工作液用该空白基质稀释成浓度为20~1.5*104ng/ml的该质控样品,该空白基质为不含该有效成分的该哺乳动物的血浆。
37.进一步地,该内标工作液的制备包括以下步骤:称量适量的柳胺酚,用第二溶剂溶解得到1mg/ml的柳胺酚储备液,将该柳胺酚储备液用第二溶剂稀释浓度为200ng/ml和50ng/ml的该内标工作液。
38.进一步地,该第一溶剂和该第二溶剂是相同的或不同的。
39.进一步地,该第一溶剂和该第二溶剂选自水、c1~c4的醇、二醇、乙腈中的一种或多种。
40.进一步地,该第一溶剂和该第二溶剂均为甲醇。
41.进一步地,该校正标示样品工作液的梯度浓度分别为400000、80000、32000、3200、800、400ng/ml。
42.进一步地,该质控工作液的梯度浓度分别为3*105、2*105、1.2*104、1200、400ng/ml。
43.进一步地,该校正标示样品的梯度浓度分别为2*104、1.6*104、4*103、1.6*103、160、40、20ng/ml。
44.进一步地,该质控样品的梯度浓度分别为1.5*104、1*104、600、60、20ng/ml。
45.进一步地,该校正标示样品和该质控样品是使用检测当天获取的该空白基质连续稀释配置而获得的。
46.进一步地,该离心的条件为8,000~15,000rpm,4~10℃条件下离心10~15min。
47.进一步地,该待测样本、校正标示样品和质控样品的体积为10~50μl。
48.进一步地,该内标工作液的体积为300~400μl。
49.进一步地,该第一上清液、该第二上清液和该第三上清液的体积为50~150μl。
50.进一步地,该第一溶剂的体积为200~400μl。
51.进一步地,该涡旋的时间为20s至2min。
52.进一步地,该待测液、该第四上清液和该第五上清液的体积为100~200μl。
53.进一步地,该离心的条件为12,000rpm,10℃条件下离心10min。
54.进一步地,该待测样本、校正标示样品和质控样品的体积为30μl。
55.进一步地,该内标工作液的体积为370μl。
56.进一步地,该第一上清液、该第二上清液和该第三上清液的体积为100μl。
57.进一步地,该第一溶剂的体积为300μl。
58.进一步地,该涡旋的时间为30s。
59.进一步地,该待测液、该第四上清液和该第五上清液的体积为150μl。
60.进一步地,该酸水溶液选自不同浓度的弱酸及其盐、弱碱及其盐、多元弱酸及其盐
中的一种或多种。
61.进一步地,该酸为甲酸、乙酸、冰乙酸、磷酸、三氟乙酸、甲酸和甲酸铵、乙酸和乙酸钠、冰乙酸和乙酸钠、乙酸和乙酸铵、冰乙酸和乙酸铵、磷酸氢二钠和磷酸二氢钠、磷酸氢二钠和磷酸二氢钾、磷酸氢二钠和柠檬酸、柠檬酸和柠檬酸钠、甘氨酸和盐酸、或者邻苯二甲酸和盐酸。
62.进一步地,该酸水溶液为0.1%至0.5%甲酸水溶液。
63.进一步地,该酸水溶液为0.1%或0.5%甲酸水溶液。
64.进一步地,该液相色谱-质谱联用的色谱条件进一步包括:柱温:26-45℃,流速:0.1-0.5ml/min;进样体积2-10μl。
65.进一步地,柱温:45℃,流速:0.3ml/min;进样体积3μl。
66.进一步地,自动进样器的温度为10℃。
67.进一步地,该自动进样器的清洗液为水、甲醇、乙腈和异丙醇以1:1:1:1比例混合的溶液。
68.进一步地,该液相色谱-质谱联用的质谱条件进一步包括:分辨率70,000;鞘气流速28au;辅助气流速15au;毛细管温度320℃;s透镜射频水平60(s-lens rf level 60);辅助气温度320℃;喷雾电压+3.4kv和/或-2.8kv。
69.进一步地,质谱仪为四极杆-静电场轨道阱高分辨质谱系统。
70.进一步地,雾化气体和干燥气体均为氮气。
71.进一步地,该质谱仪的检测条件为选择性离子检测。
72.根据本发明的另一个方面,本发明提供了一种根据上述方法在构建检测试剂盒或检测装置中的用途,该检测试剂盒或该检测装置用于检测血液中扶正化瘀制剂有效成分浓度,其中该有效成分为以下中的一种或多种:五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素。
73.应用本发明的技术方案,本发明首先利用uhplc,选择合适的有机溶剂及合适的比例,可使血液中的待测组分在流动相中分离,再通过质谱检测仪进行检测,且分析方法灵敏和快捷,以利于进行血液中五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素的测定。
附图说明
74.为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,还可以根据这些附图获得其他的附图,而并不超出本发明要求保护的范围。
75.图1是本发明实施例一中五味子醇乙-空白(black)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
76.图2是本发明实施例一中五味子醇乙-qc0样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
77.图3是本发明实施例一中五味子醇乙-残留效应(carryover blank)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
78.图4是本发明实施例一中五味子醇乙-lloq样品的谱图。纵坐标:相对丰度(%);横
坐标:时间(min)。
79.图5是本发明实施例一中五味子醇乙-uloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
80.图6是本发明实施例二中五味子甲素-空白(black)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
81.图7是本发明实施例二中五味子甲素-qc0样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
82.图8是本发明实施例二中五味子甲素-残留效应(carryover blank)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
83.图9是本发明实施例二中五味子甲素-lloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
84.图10是本发明实施例二中五味子甲素-uloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
85.图11是本发明实施例三中丹酚酸d-溶剂(reagent)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
86.图12是本发明实施例三中丹酚酸d空白(bk)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
87.图13是本发明实施例三中丹酚酸d-qc0样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
88.图14是本发明实施例三中丹酚酸d-残留效应(carryover blank)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
89.图15是本发明实施例三中丹酚酸d-lloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
90.图16是本发明实施例三中丹酚酸d-uloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
91.图17是本发明实施例四中咖啡酸-溶剂(reagent)的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
92.图18是本发明实施例四中咖啡酸空白(bk)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
93.图19是本发明实施例四中咖啡酸-qc0样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
94.图20是本发明实施例四中咖啡酸-残留效应(carryover blank)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
95.图21是本发明实施例四中咖啡酸-lloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
96.图22是本发明实施例四中咖啡酸-uloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
97.图23是本发明实施例四中染料木素-溶剂(reagent)的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
98.图24是本发明实施例四中染料木素空白(bk)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
99.图25是本发明实施例四中染料木素-qc0样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
100.图26是本发明实施例四中染料木素-残留效应(carryover blank)样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
101.图27是本发明实施例四中染料木素-lloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
102.图28是本发明实施例四中染料木素-uloq样品的谱图。纵坐标:相对丰度(%);横坐标:时间(min)。
具体实施方式
103.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
104.在本发明的描述中,需要说明的是,术语“上”、“下”、“底”、“内”、“外”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。此外,术语“第一”、“第二”等仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”等的特征可以明示或隐含地包括一个或者更多个该特征。
105.需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
106.以下结合具体实施例对本发明作进一步详细描述,这些实施例不能理解为限制本发明所要求保护的范围。
107.本发明公开了一种测定血液中扶正化瘀制剂五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和/或染料木素浓度的方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。
108.除特别指出,本发明提供的技术方案中所用药品、试剂、仪器均可由常规渠道或市场购得。
109.本发明提供了一种测定血液中扶正化瘀制剂有效成分浓度的方法,该方法包括以下步骤:
110.(1)待测样本预处理,获取待测液;
111.(2)有效成分标准曲线制备;以及
112.(3)液相色谱-质谱联用检测该待测液中该有效成分浓度,
113.其中,该液相色谱-质谱联用的色谱条件为:色谱柱:acquity uplc hss t3(2.1mm
×
100mm,1.8μm),流动相:水相(a)为酸水溶液,有机相(b)为甲醇或乙腈;洗脱方式:采用以
下梯度程序洗脱:0-12min,5%b;12-14min,95%b;14-16min,5%b;
114.其中,该液相色谱-质谱联用的质谱条件为:电喷雾离子源(esi),采用正负离子模式检测,质量扫描范围m/z 100-800;
115.其中,该有效成分为以下中的一种或多种:五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素。
116.在一种优选的实施方式中,该待测样本为哺乳动物的血浆。
117.在一种优选的实施方式中,该哺乳动物为大鼠、小鼠或犬。
118.在一种优选的实施方式中,该预处理包括以下步骤:
119.(1)吸取该待测样本,添加至第一96孔板中;
120.(2)在该第一96孔板中添加内标工作液;
121.(3)封板后涡旋混匀并离心,得到第一上清液;
122.(4)吸取该第一上清液,添加至第二96孔板中;
123.(5)在该第二96孔板中添加第一溶剂;以及
124.(6)封板后涡旋混匀并离心,得到该待测液。
125.在一种优选的实施方式中,该有效成分标准曲线制备包括以下步骤:
126.(1)分别吸取校正标示样品和质控样品,分别添加至第一96孔板中;
127.(2)在该第一96孔板中分别添加内标工作液;
128.(3)封板后涡旋混匀并离心,得到第二上清液和第三上清液;
129.(4)分别吸取该第二上清液和第三上清液,分别添加至第二96孔板中;
130.(5)在该第二96孔板中分别添加第一溶剂;
131.(6)封板后涡旋混匀并离心,得到第四上清液和第五上清液;
132.(7)对该第四上清液和该第五上清液进行该液相色谱-质谱联用分析,得到数据;以及
133.(8)使用内标法对该数据进行处理,分别以该有效成分的浓度为横坐标,该有效成分和柳胺酚的峰面积比值为纵坐标,以1/x2为权重系数,利用加权最小二乘法进行线性回归,得到该有效成分的标准曲线。
134.在定量过程中,以五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和/或染料木素为对照品,并以柳胺酚为内标,采用内标法准确、灵敏地监测,实现对五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和/或染料木素的定量检测。
135.在一种优选的实施方式中,该校正标示样品的制备包括以下步骤:
136.(1)称量适量的有效成分,用第二溶剂溶解得到1mg/ml的有效成分储备液,将该有效成分储备液用第二溶剂稀释成梯度浓度400~4*105ng/ml的校正标示样品工作液;以及
137.(2)将该校正标示样品工作液用空白基质稀释成梯度浓度为20~2*104ng/ml的该校正标示样品,该空白基质为不含该有效成分的该哺乳动物的血浆;
138.在一种优选的实施方式中,该质控样品的制备包括以下步骤:
139.(1)称量适量的有效成分,用第二溶剂溶解得到1mg/ml的有效成分储备液,将该有效成分储备液用第二溶剂稀释成梯度浓度400~3*105ng/ml的质控工作液;以及
140.(2)将该质控工作液用该空白基质稀释成浓度为20~1.5*104ng/ml的该质控样品,该空白基质为不含该有效成分的该哺乳动物的血浆。
141.在一种优选的实施方式中,该内标工作液的制备包括以下步骤:称量适量的柳胺酚,用第二溶剂溶解得到1mg/ml的柳胺酚储备液,将该柳胺酚储备液用第二溶剂稀释浓度为200ng/ml和50ng/ml的该内标工作液。
142.在一种优选的实施方式中,该第一溶剂和该第二溶剂是相同的或不同的。
143.在一种优选的实施方式中,该第一溶剂和该第二溶剂选自水、c1~c4的醇、二醇、乙腈中的一种或多种。
144.在一种优选的实施方式中,该第一溶剂和该第二溶剂均为甲醇。
145.在一种优选的实施方式中,该校正标示样品工作液的梯度浓度分别为400000、80000、32000、3200、800、400ng/ml。
146.在一种优选的实施方式中,该质控工作液的梯度浓度分别为3*105、2*105、1.2*104、1200、400ng/ml。
147.在一种优选的实施方式中,该校正标示样品的梯度浓度分别为2*104、1.6*104、4*103、1.6*103、160、40、20ng/ml。
148.在一种优选的实施方式中,该质控样品的梯度浓度分别为1.5*104、1*104、600、60、20ng/ml。
149.在一种优选的实施方式中,该校正标示样品和该质控样品是使用检测当天获取的该空白基质连续稀释配置而获得的。
150.在一种优选的实施方式中,该离心的条件为8,000~15,000rpm,4~10℃条件下离心10~15min。
151.在一种优选的实施方式中,该待测样本、校正标示样品和质控样品的体积为10~50μl。
152.在一种优选的实施方式中,该内标工作液的体积为300~400μl。
153.在一种优选的实施方式中,该第一上清液、该第二上清液和该第三上清液的体积为50~150μl。
154.在一种优选的实施方式中,该第一溶剂的体积为200~400μl。
155.在一种优选的实施方式中,该涡旋的时间为20s至2min。
156.在一种优选的实施方式中,该待测液、该第四上清液和该第五上清液的体积为100~200μl。
157.在一种优选的实施方式中,该离心的条件为12,000rpm,10℃条件下离心10min。
158.在一种优选的实施方式中,该待测样本、校正标示样品和质控样品的体积为30μl。
159.在一种优选的实施方式中,该内标工作液的体积为370μl。
160.在一种优选的实施方式中,该第一上清液、该第二上清液和该第三上清液的体积为100μl。
161.在一种优选的实施方式中,该第一溶剂的体积为300μl。
162.在一种优选的实施方式中,该涡旋的时间为30s。
163.在一种优选的实施方式中,该待测液、该第四上清液和该第五上清液的体积为150μl。
164.在一种优选的实施方式中,该酸水溶液选自不同浓度的弱酸及其盐、弱碱及其盐、多元弱酸及其盐中的一种或多种。
165.在一种优选的实施方式中,该酸为甲酸、乙酸、冰乙酸、磷酸、三氟乙酸、甲酸和甲酸铵、乙酸和乙酸钠、冰乙酸和乙酸钠、乙酸和乙酸铵、冰乙酸和乙酸铵、磷酸氢二钠和磷酸二氢钠、磷酸氢二钠和磷酸二氢钾、磷酸氢二钠和柠檬酸、柠檬酸和柠檬酸钠、甘氨酸和盐酸、或者邻苯二甲酸和盐酸。
166.为了使色谱峰得到更好的分离,保留时间适宜,且重现性和稳定性更优,在一种优选的实施方式中,该酸水溶液为0.1%至0.5%甲酸水溶液。
167.在一种优选的实施方式中,该酸水溶液为0.1%或0.5%甲酸水溶液。
168.在一种优选的实施方式中,该液相色谱-质谱联用的色谱条件进一步包括:柱温:26-45℃,流速:0.1-0.5ml/min;进样体积2-10μl。
169.为了获得更好的色谱峰分离效果且具有较低的柱压,在一种优选的实施方式中,柱温:45℃,流速:0.3ml/min;进样体积3μl。
170.在一种优选的实施方式中,自动进样器的温度为10℃。
171.在一种优选的实施方式中,该自动进样器的清洗液为水、甲醇、乙腈和异丙醇以1:1:1:1比例混合的溶液。
172.在一种优选的实施方式中,该液相色谱-质谱联用的质谱条件进一步包括:分辨率70,000;鞘气流速28au;辅助气流速15au;毛细管温度320℃;s透镜射频水平60;辅助气温度320℃;喷雾电压+3.4kv和/或-2.8kv。
173.在一种优选的实施方式中,质谱仪为四极杆-静电场轨道阱高分辨质谱系统。
174.在一种优选的实施方式中,雾化气体和干燥气体均为氮气。
175.在一种优选的实施方式中,该质谱仪的检测条件为选择性离子检测。
176.本发明通过大量实验筛选出最佳的流动相组成、梯度洗脱方式、检测波长、色谱柱,柱温等分析条件,经多次实验验证表明,本发明提供的检测方法,可以全面、客观、准确的检测和评价血液中扶正化瘀制剂五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和/或染料木素的含量。
177.根据本发明的另一个方面,本发明提供了一种根据上述方法在构建检测试剂盒或检测装置中的用途,该检测试剂盒或该检测装置用于检测血液中扶正化瘀制剂有效成分浓度,其中该有效成分为以下中的一种或多种:五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素。
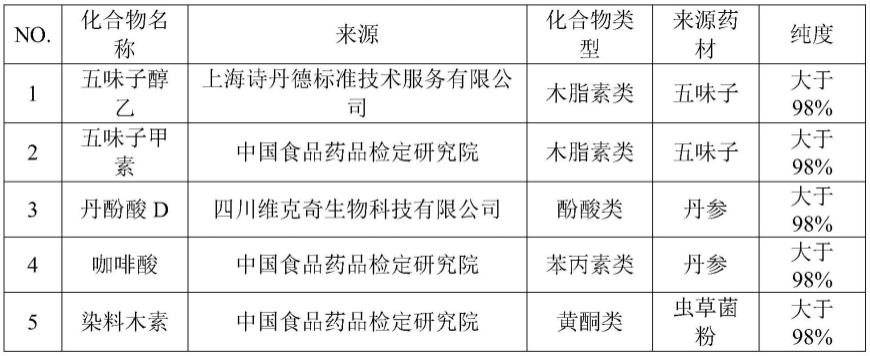
178.其中五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素来源、化合物类型与纯度如表1所示。
179.表1五味子醇乙、五味子甲素、丹酚酸d、咖啡酸和染料木素来源、化合物类型与纯度
[0180][0181]
实施例
[0182]
实施例一
[0183]
1、样品的配置
[0184]
储备液的配置:五味子醇乙和柳胺酚(内标,来源于sigma-aldrich,纯度大于98%)各取1.00mg,并且分别用甲醇溶解,配置得到五味子醇乙储备液和柳胺酚储备液,≤-15℃密闭,遮光保存,其中柳胺酚作为内标,如表2所示。
[0185]
表2储备液的配置方法
[0186]
溶液浓度容器溶剂储存条件五味子醇乙储备液1.00mg/ml棕色玻璃瓶甲醇≤-15℃,密闭避光柳胺酚储备液1.00mg/ml棕色玻璃瓶甲醇≤-15℃,密闭避光
[0187]
校正标示样品工作液的配置:将五味子醇乙储备液用甲醇溶液稀释成梯度浓度400~4*105ng/ml的校正标示样品工作液,如表3所示。
[0188]
表3校正标示样品工作溶液的配置方法
[0189][0190][0191]
质控工作液的配置:将五味子醇乙储备液用甲醇溶液稀释成梯度浓度400~3*105ng/ml的质控工作液,如表4所示。
[0192]
表4质控工作液的配置方法
[0193]
分析物源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)五味子醇乙1000000300700300000五味子醇乙300000300150200000
五味子醇乙3000002048012000五味子醇乙12000504501200五味子醇乙1200150300400
[0194]
内标工作液的配置:将柳胺酚储备液用甲醇溶液稀释成浓度为200、50ng/ml的内标工作液,如表5所示。
[0195]
表5内标工作液的配置方法
[0196]
源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)10000005.002500020020010030050.0
[0197]
neat溶液的配置:将质控工作液和内标工作液用等体积的甲醇溶液稀释成neat溶液,如表6所示。
[0198]
表6 neat溶液的配置方法
[0199][0200]
上述校正标示样品工作液、质控工作液、内标工作液以及回收率和基质效应纯溶液(简称neat溶液),只要终浓度不变,稀释过程/体积可以改变。
[0201]
校正标示样品的配置:将校正标示样品工作液用空白基质稀释成浓度为2*104、1.6*104、4*103、1.6*103、160、40、20ng/ml的校正标示样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含五味子醇乙,如表7所示。
[0202]
表7校正标示样品的配置方法
[0203][0204]
质控样品的配置:将质控工作液用空白基质稀释成浓度为1.5*104、1*104、600、60、20ng/ml的质控样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含五味子醇乙,如表8所示。
[0205]
表8质控样品的配置方法
[0206]
工作液浓度(ng/ml)工作液体积(μl)空白基质体积(μl)质控样品浓度(ng/ml)300000101901500020000010190100001200010190600120010190604001019020
[0207]
2、uhplc-ms/ms分析测定过程的工作条件
[0208]
(1)色谱条件:
[0209]
色谱柱:acquity uplc hss t3(2.1mm
×
100mm,1.8μm),柱温为45℃;
[0210]
自动进样器温度:10℃;
[0211]
流动相a:0.1%甲酸水溶液;
[0212]
流动相b:甲醇溶液;
[0213]
进样器清洗液:水:甲醇:乙腈:异丙醇=1:1:1:1;
[0214]
洗脱方式:梯度洗脱;
[0215]
泵梯度:第0~12min,流动相b的体积分数为5%;第12~14min,流动相b的体积分数为95%;第14~16min,流动相b的体积分数为5%;
[0216]
进样体积:3μl;
[0217]
流速:0.3ml/min;
[0218]
运行时间:16min;
[0219]
(2)质谱条件:
[0220]
质谱仪:thermo scientific q exactive四极杆-静电场轨道阱高分辨质谱系统
[0221]
离子源:hesi电喷雾电离;
[0222]
离子化模式:正负离子模式;
[0223]
检测模式:sim单离子检测(选择性离子检测),质量扫描范围m/z100-800;
[0224]
分辨率70,000;鞘气流速28au;辅助气流速15au;毛细管温度320℃;s透镜射频水平60;辅助气温度320℃;喷雾电压+3.4kv和/或-2.8kv。
[0225]
3、样品处理
[0226]
吸取30μl的校正标示样品、质控样品,再转移至96孔板中,然后再分别加入370μl的50ng/ml内标工作液,封板后涡旋混匀,在10℃,12,000rpm条件下离心10min。离心后取上清液100μl至96孔板中,并加入300μl的甲醇溶液。然后再密封孔板,进行低速涡旋混匀30s,取150μl在超高效液相质谱联用仪(uhplc-ms/ms)上进行分析。
[0227]
4、直线回归模型的建立
[0228]
按上述色谱和质谱条件对校正标示样品、质控样品进行检测,以五味子醇乙比内标柳胺酚的色谱峰面积比为纵坐标,以犬血浆中五味子醇乙的浓度为横坐标,用加权(权重为1/x2)最小二乘法以犬血浆中五味子醇乙的浓度(x)与色谱峰面积比(y)进行线性回归,所构建的线性回归方程(y=ax+b)即为标准曲线。结果表明,五味子醇乙的校正曲线范围20.0ng/ml-20000ng/ml内线性良好。
[0229]
5、定量分析
[0230]
取30μl待测样品按照上述的样品处理步骤的方法处理,并按样品处理步骤所得的
标准曲线方程计算,得到待测样品中分析物五味子醇乙的浓度。
[0231]
6、测试方法的评价
[0232]
制备空白(bk)样品、qc0样品、uloq without is样品用来相互校正,减少干扰性。吸取30μl的空白基质放入空白样品孔内,空白样品中无分析物,无内标;吸取30μl空白基质中放入qc0样品孔内;吸取30μl最高定量限不含内标的空白基质放入uloq without is样品孔内;在30μl空白基质中加入370μl内标工作液形成qc0样品;空白(bk)样品、uloq without is样品中加入370μl甲醇溶液;空白(bk)样品、qc0样品、uloq without is样品在稀释混合后涡旋离心取上清液100μl,再加入300μl的甲醇溶液,涡旋混匀后取150μl进样分析。
[0233]
制备基质效应样品和回收率样品用来评价该方法的基质效应和回收率。取单个供体(n≥6)的30μl的空白基质,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的基质效应为93.0~100%。
[0234]
取单个供体的空白基质,再取30μl的多个供体的混合的空白基质形成回收率样品,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的五味子醇乙的回收率为100%。
[0235]
实施例二
[0236]
1、样品的配置
[0237]
储备液的配置:对五味子甲素和柳胺酚(内标,来源于sigma-aldrich,纯度大于98%)各取1.00mg,并且分别用甲醇溶解,配置得到五味子甲素储备液、柳胺酚储备液,≤-15℃密闭,遮光保存,其中柳胺酚作为内标,如表9所示。
[0238]
表9储备液的配置方法
[0239]
溶液浓度容器溶剂储存条件五味子甲素储备液1.00mg/ml棕色玻璃瓶甲醇≤-15℃,密闭避光柳胺酚储备液1.00mg/ml棕色玻璃瓶甲醇≤-15℃,密闭避光
[0240]
校正标示样品工作液的配置:将五味子甲素储备液用甲醇溶液稀释成梯度浓度400~4*105ng/ml的校正标示样品工作液,如表10所示。
[0241]
表10校正标示样品工作液的配置方法
[0242]
分析物源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)五味子甲素1000000200300400000五味子甲素40000010040080000五味子甲素4000004046032000五味子甲素80000204803200五味子甲素3200125375800五味子甲素320075525400
[0243]
质控工作液的配置:将五味子甲素储备液用甲醇溶液稀释成梯度浓度400~3*105ng/ml的质控工作液,如表11所示。
[0244]
表11质控工作液的配置方法
[0245]
分析物源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)五味子甲素1000000300700300000
五味子甲素300000300150200000五味子甲素3000002048012000五味子甲素12000504501200五味子甲素1200150300400
[0246]
内标工作液的配置:将柳胺酚储备液用甲醇溶液稀释成浓度为200、50ng/ml的内标工作液,如表12所示。
[0247]
表12内标工作液的配置方法
[0248]
源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)10000005.002500020020010030050.0
[0249]
neat溶液的配置:将质控工作液和内标工作液用等体积的甲醇溶液稀释成neat溶液,如表13所示。
[0250]
表13 neat溶液的配置方法
[0251][0252]
上述校正标示样品工作液、质控工作液、内标工作液以及回收率和基质效应纯溶液(简称neat溶液),只要终浓度不变,稀释过程/体积可以改变。
[0253]
校正标示样品的配置:将校正标示样品工作液用空白基质稀释成浓度为2*104、1.6*104、4*103、1.6*103、160、40、20ng/ml的校正标示样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含五味子甲素,如表14所示。
[0254]
表14校正标示样品的配置方法
[0255][0256]
质控样品的配置:将质控工作液用空白基质稀释成浓度为1.5*104、1*104、600、60、20ng/ml的质控样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含五味子甲素,如表15所示。
[0257]
表15质控样品的配置方法
[0258]
工作液浓度(ng/ml)工作液体积(μl)空白基质体积(μl)质控样品浓度(ng/ml)300000101901500020000010190100001200010190600120010190604001019020
[0259]
2、uhplc-ms/ms分析测定过程的工作条件
[0260]
(1)色谱条件:
[0261]
色谱柱:acquity uplc hss t3(2.1mm
×
100mm,1.8μm),柱温为45℃;
[0262]
自动进样器温度:10℃;
[0263]
流动相a:0.5%甲酸水溶液;
[0264]
流动相b:乙腈溶液;
[0265]
进样器清洗液:水:甲醇:乙腈:异丙醇=1:1:1:1;
[0266]
洗脱方式:梯度洗脱;
[0267]
泵梯度:第0~12min,流动相b的体积分数为5%;第12~14min,流动相b的体积分数为95%;第14~16min,流动相b的体积分数为5%;
[0268]
进样体积:3μl;
[0269]
流速:0.3ml/min;
[0270]
运行时间:16min;
[0271]
(2)质谱条件:
[0272]
质谱仪:thermo scientific q exactive四极杆-静电场轨道阱高分辨质谱系统
[0273]
离子源:hesi电喷雾电离;
[0274]
离子化模式:正负离子模式;
[0275]
检测模式:sim单离子检测(选择性离子检测),质量扫描范围m/z100-800;
[0276]
分辨率70,000;鞘气流速28au;辅助气流速15au;毛细管温度320℃;s透镜射频水平60;辅助气温度320℃;喷雾电压+3.4kv和/或-2.8kv。
[0277]
3、样品处理
[0278]
吸取30μl的校正标示样品、质控样品,再转移至96孔板中,然后再分别加入370μl的50ng/ml内标工作液,封板后涡旋混匀,在10℃,12,000rpm条件下离心10min。离心后取上清液100μl至96孔板中,并加入300μl的甲醇溶液。然后再密封孔板,进行低速涡旋混匀30s,取150μl在高效液相质谱联用仪(uhplc-ms/ms)上进行分析。
[0279]
4、直线回归模型的建立
[0280]
按上述色谱和质谱条件对校正标示样品、质控样品进行检测。以以五味子甲素比内标柳胺酚的色谱峰面积比为纵坐标,以犬血浆中五味子甲素的浓度为横坐标,用加权(权重为1/x2)最小二乘法以犬血浆中五味子甲素的浓度(x)与色谱峰面积比(y)进行线性回归,所构建的线性回归方程(y=ax+b)即为标准曲线。结果表明,五味子甲素的校正曲线范围20.0ng/ml-20000ng/ml内线性良好。
[0281]
5、定量分析
[0282]
取30μl待测样品按照上述的样品处理步骤的方法处理,并按样品处理步骤所得的标准曲线方程计算,得到待测样品中分析物五味子甲素的浓度。
[0283]
6、测试方法的评价
[0284]
制备空白(bk)样品、qc0样品、uloq without is样品用来相互校正,减少干扰性。吸取30μl的空白基质放入空白样品孔内,空白样品中无分析物,无内标;吸取30μl空白基质中放入qc0样品孔内;吸取30μl最高定量限不含内标的空白基质放入uloq without is样品孔内;在30μl空白基质中加入370μl内标工作液形成qc0样品;空白(bk)样品、uloq without is样品中加入370μl甲醇溶液;空白(bk)样品、qc0样品、uloq without is样品在稀释混合后涡旋离心取上清液100μl,再加入300μl的甲醇溶液,涡旋混匀后取150μl进样分析。
[0285]
制备基质效应样品和回收率样品用来评价该方法的基质效应和回收率。取单个供体(n≥6)的30μl的空白基质,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的基质效应为93.0~100%。
[0286]
取单个供体的空白基质,再取30μl的多个供体的混合的空白基质形成回收率样品,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的五味子甲素的回收率为100%。
[0287]
实施例三
[0288]
1、样品的配置
[0289]
储备液的配置:丹酚酸d和柳胺酚(内标,来源于sigma-aldrich,纯度大于98%)各取4.00mg,并且分别溶于甲醇中,配置得到丹酚酸d储备液和柳胺酚储备液。≤
–
15℃密闭,遮光保存,其中柳胺酚作为内标,如表16所示。
[0290]
表16储备液的配置方法
[0291][0292][0293]
校正标示样品工作液的配置:将丹酚酸d储备液用甲醇溶液稀释成梯度浓度400~4*105ng/ml的校正标示样品工作液,如表17所示。
[0294]
表17校正标示样品工作液的配置方法
[0295]
分析物源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)丹酚酸d1000000200300400000丹酚酸d40000010040080000丹酚酸d4000004046032000丹酚酸d80000204803200丹酚酸d3200125375800丹酚酸d320075525400
[0296]
质控工作液的配置:将丹酚酸d储备液用甲醇溶液稀释成梯度浓度400~3*105ng/
ml的质控工作液,如表18所示。
[0297]
表18质控工作液的配置方法
[0298]
分析物源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)丹酚酸d1000000300700300000丹酚酸d300000300150200000丹酚酸d3000002048012000丹酚酸d12000504501200丹酚酸d1200150300400
[0299]
内标工作液的配置:将柳胺酚储备液用甲醇溶液稀释成浓度为200、50ng/ml的内标工作液,如表19所示。
[0300]
表19内标工作液的配置方法
[0301]
源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)10000005.002500020020010030050.0
[0302]
neat溶液的配置:将质控溶液和内标工作液用等体积的甲醇溶液稀释成neat溶液,如表20所示。
[0303]
表20 neat溶液的配置方法
[0304][0305]
上述校正标示样品工作液、质控工作液、内标工作液以及回收率和基质效应纯溶液(简称neat溶液),只要终浓度不变,稀释过程/体积可以改变。
[0306]
校正标示样品的配置:将校正标示样品工作液用空白基质稀释成浓度为2*104、1.6*104、4*103、1.6*103、160、40、20ng/ml的校正标示样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含丹酚酸d和柳胺酚,如表21所示。
[0307]
表21校正标示样品的配置方法
[0308][0309]
质控样品的配置:将质控工作液用空白基质稀释成浓度为1.5*104、1*104、600、60、20ng/ml的质控样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含丹酚酸d和柳胺酚,如表22所示。
[0310]
表22质控样品的配置方法
[0311][0312][0313]
2、uhplc-ms/ms分析测定过程的工作条件
[0314]
(1)uhplc条件:
[0315]
色谱柱:acquity uplc hss t3(2.1mm
×
100mm,1.8μm),沃特世;
[0316]
柱温:45℃;
[0317]
流动相a:0.1%甲酸水溶液;
[0318]
流动相b:甲醇溶液;
[0319]
泵梯度:第0~12min,流动相b的体积分数为5%;第12~14min,流动相b的体积分数为95%;第14~16min,流动相b的体积分数为5%;
[0320]
进样体积:3.00μl;
[0321]
流速:0.3ml/min;
[0322]
运行时间:16min;
[0323]
自动进样器温度:10℃;
[0324]
进样器清洗液:水:甲醇:乙腈:异丙醇=1:1:1:1;
[0325]
(2)质谱条件:
[0326]
质谱仪:thermo scientific q exactive四极杆-静电场轨道阱高分辨质谱系统;
[0327]
离子源:hesi;
[0328]
离子化模式:正负离子模式;
[0329]
检测模式:sim单离子检测(选择性离子检测),质量扫描范围m/z100-800;
[0330]
分辨率70,000;鞘气流速28au;辅助气流速15au;毛细管温度320℃;s透镜射频水平60;辅助气温度320℃;喷雾电压+3.4kv和/或-2.8kv。
[0331]
3、样品处理
[0332]
吸取30μl的校正标示样品、质控样品,再转移至96孔板中,然后再分别加入370μl的50ng/ml内标工作液,封板后涡旋混匀,在10℃,12,000rpm条件下离心10min。离心后取上清液100μl至96孔板中,并加入300μl的甲醇溶液。然后再密封孔板,进行低速涡旋混匀30s,取150μl在超高效液相质谱联用仪(uhplc-ms/ms)上进行分析。
[0333]
4、直线回归模型的建立
[0334]
按上述色谱和质谱条件对校正标示样品、质控样品进行检测。以丹酚酸d比内标柳胺酚的色谱峰面积比为纵坐标,以犬血浆中丹酚酸d的浓度为横坐标,用加权(权重为1/x2)最小二乘法以犬血浆丹酚酸d的浓度(x)与色谱峰面积比(y)进行线性回归,所构建的线性回归方程(y=ax+b)即为标准曲线。结果表明,丹酚酸d的校正曲线范围在20ng/ml-2*104ng/ml内线性良好。
[0335]
5、定量分析
[0336]
取试验样品按照上述的样品处理步骤的方法处理,并按样品处理步骤所得的标准曲线方程计算,得到待测样品中分析物丹酚酸d的浓度。
[0337]
6、测试方法的评价
[0338]
制备空白(bk)样品、qc0样品、uloq without is样品用来相互校正,减少干扰性。吸取30μl的空白基质放入空白样品孔内,空白样品中无分析物,无内标;吸取30μl空白基质中放入qc0样品孔内;吸取30μl最高定量限不含内标的空白基质放入uloq without is样品孔内;在30μl空白基质中加入370μl内标工作液形成qc0样品;空白(bk)样品、uloq without is样品中加入370μl甲醇溶液;空白(bk)样品、qc0样品、uloq without is样品在稀释混合后涡旋离心取上清液100μl,再加入300μl的甲醇溶液,涡旋混匀后取150μl进样分析。
[0339]
制备基质效应样品和回收率样品用来评价该方法的基质效应和回收率。取单个供体(n≥6)的30μl的空白基质,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的基质效应为100~110%。
[0340]
取单个供体的空白基质,再取30μl的多个供体的混合的空白基质形成回收率样品,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的丹酚酸d回收率为:105.17%。
[0341]
实施例四
[0342]
1、样品的配置
[0343]
储备液的配置:对咖啡酸、染料木素和柳胺酚(内标,来源于sigma-aldrich,纯度大于98%)各取4.00mg,并且分别溶于甲醇中,配置得到咖啡酸储备液、染料木素储备液和柳胺酚储备液。≤
–
15℃密闭,遮光保存,其中柳胺酚作为内标,如表23所示。
[0344]
表23储备液的配置方法
[0345]
溶液浓度容器溶剂储存条件
咖啡酸储备液1.00mg/ml棕色玻璃瓶甲醇≤
–
15℃,密闭避光染料木素储备液1.00mg/ml棕色玻璃瓶甲醇≤
–
15℃,密闭避光柳胺酚储备液1.00mg/ml棕色玻璃瓶甲醇≤
–
15℃,密闭避光
[0346]
校正标示样品工作液的配置:将咖啡酸储备液和染料木素储备液分别用甲醇溶液稀释成梯度浓度400~4*105ng/ml的校正标示样品工作液,如表24所示。
[0347]
表24校正标示样品工作液的配置方法
[0348][0349]
质控工作液的配置:将咖啡酸储备液和染料木素储备液分别用甲醇溶液稀释成梯度浓度400~3*105ng/ml的质控工作液,如表25所示。
[0350]
表25质控工作液的配置方法
[0351][0352]
内标工作液的配置:将柳胺酚储备液用甲醇溶液稀释成浓度为200、50ng/ml的内标工作液,如表26所示。
[0353]
表26内标工作液的配置方法
[0354]
源溶液浓度(ng/ml)源溶液体积(μl)溶剂体积(μl)终浓度(ng/ml)10000005.002500020020010030050.0
[0355]
neat溶液的配置:将质控溶液和内标工作液用等体积的甲醇溶液稀释成neat溶液,如表27所示。
[0356]
表27 neat溶液的配置方法
[0357][0358]
上述校正标示样品工作液、质控工作液、内标工作液以及回收率和基质效应纯溶液(简称neat溶液),只要终浓度不变,稀释过程/体积可以改变。
[0359]
校正标示样品的配置:将校正标示样品工作液用空白基质稀释成浓度为2*104、1.6*104、4*103、1.6*103、160、40、20ng/ml的校正标示样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含咖啡酸、染料木素和柳胺酚,如表28所示。
[0360]
表28校正标示样品的配置方法
[0361][0362]
质控样品的配置:将质控工作液用空白基质稀释成浓度为1.5*104、1*104、600、60、20ng/ml的质控样品,空白基质为不含分析物与内标的犬血浆,在本实施例中指不含咖啡酸、染料木素和柳胺酚,如表29所示。
[0363]
表29质控样品的配置方法
[0364]
工作液浓度(ng/ml)工作液体积(μl)空白基质体积(μl)质控样品浓度(ng/ml)300000/3000001019015000/15000200000/2000001019010000/1000012000/1200010190600/6001200/12001019060/60400/4001019020/20
[0365]
2、uhplc-ms/ms分析测定过程的工作条件
[0366]
(1)uhplc条件:
[0367]
色谱柱:acquity uplc hss t3(2.1mm
×
100mm,1.8μm),沃特世;
[0368]
柱温:45℃;
[0369]
流动相a:0.1%甲酸水溶液;
[0370]
流动相b:乙腈;
[0371]
泵梯度:第0~12min,流动相b的体积分数为5%;第12~14min,流动相b的体积分数为95%;第14~16min,流动相b的体积分数为5%;
[0372]
进样体积:3.00μl;
[0373]
流速:0.3ml/min;
[0374]
运行时间:16min;
[0375]
自动进样器温度:10℃;
[0376]
进样器清洗液:水:甲醇:乙腈:异丙醇=1:1:1:1;
[0377]
(2)质谱条件:
[0378]
质谱仪:thermo scientific q exactive四极杆-静电场轨道阱高分辨质谱系统;
[0379]
离子源:hesi;
[0380]
离子化模式:正负离子模式;
[0381]
检测模式:sim单离子检测(选择性离子检测),质量扫描范围m/z100-800;
[0382]
分辨率70,000;鞘气流速28au;辅助气流速15au;毛细管温度320℃;s透镜射频水平60;辅助气温度320℃;喷雾电压+3.4kv和/或-2.8kv。
[0383]
3、样品处理
[0384]
吸取30μl的校正标示样品、质控样品,再转移至96孔板中,然后再分别加入370μl的50ng/ml内标工作液,封板后涡旋混匀,在10℃,12,000rpm条件下离心10min。离心后取上清液100μl至96孔板中,并加入300μl的甲醇溶液。然后再密封孔板,进行低速涡旋混匀30s,取150μl在超高效液相质谱联用仪(uhplc-ms/ms)上进行分析。
[0385]
4、直线回归模型的建立
[0386]
按上述色谱和质谱条件对校正标示样品、质控样品进行检测。以咖啡酸比内标柳胺酚、染料木素比内标柳胺酚的色谱峰面积比为纵坐标,以犬血浆中咖啡酸、染料木素的浓度为横坐标,用加权(权重为1/x2)最小二乘法以犬血浆咖啡酸、染料木素的浓度(x)与色谱峰面积比(y)进行线性回归,所构建的线性回归方程(y=ax+b)即为标准曲线。结果表明,咖啡酸、染料木素的校正曲线范围在20ng/ml-2*104ng/ml内线性良好。
[0387]
5、定量分析
[0388]
取试验样品按照上述的样品处理步骤的方法处理,并按样品处理步骤所得的标准曲线方程计算,得到待测样品中分析物咖啡酸、染料木素的浓度。
[0389]
6、测试方法的评价
[0390]
制备空白(bk)样品、qc0样品、uloq without is样品用来相互校正,减少干扰性。吸取30μl的空白基质放入空白样品孔内,空白样品中无分析物,无内标;吸取30μl空白基质中放入qc0样品孔内;吸取30μl最高定量限不含内标的空白基质放入uloq without is样品孔内;在30μl空白基质中加入370μl内标工作液形成qc0样品;空白(bk)样品、uloq without is样品中加入370μl甲醇溶液;空白(bk)样品、qc0样品、uloq without is样品在稀释混合后涡旋离心取上清液100μl,再加入300μl的甲醇溶液,涡旋混匀后取150μl进样分析。
[0391]
制备基质效应样品和回收率样品用来评价该方法的基质效应和回收率。取单个供体(n≥6)的30μl的空白基质,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法中咖啡酸的基质效应为92.37%
±
2.42,该方法中染料木素的基质效应为99.86%
±
0.93。
[0392]
取单个供体的空白基质,再取30μl的多个供体的混合的空白基质形成回收率样品,并加入370μl甲醇溶液,涡旋离心后取上清液100μl,再加入50μl的neat溶液和250μl的甲醇溶液,涡旋混匀后取150μl进样分析。分析得到该方法的咖啡酸回收率为:95.48%;染料木素的回收率为:98.97%。
[0393]
本发明的优点:uhplc-ms/ms分析测定过程的工作条件包括uhplc和色谱条件,通过色谱柱、溶解液、流动相、保留时间等工艺条件的优化,使得整个测试过程更加可行,测量结果增加准确,减少了残留,提高了准确度以及回收率高,减小了基质效应以及干扰性。
[0394]
以上所述的仅是本发明的实施例,方案中公知的具体结构及特性等常识在此未作过多描述。应当指出,对于本领域的技术人员来说,在不脱离本发明的前提下,还可以作出若干变形和改进,这些也应该视为本发明的保护范围,这些都不会影响本发明实施的效果和专利的实用性。本发明要求的保护范围应当以其权利要求的内容为准,说明书中的具体实施方式等记载可以用于解释权利要求的内容。
[0395]
以上对本发明实施例进行了详细介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明仅用于帮助理解本发明的方法及其核心思想。同时,本领域技术人员依据本发明的思想,基于本发明的具体实施方式及应用范围上做出的改变或变形之处,都属于本发明保护的范围。综上所述,本说明书内容不应理解为对本发明的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1