一种海洋沉积物中有机碳含量的测定方法与流程
1.本发明涉及材料分析测试技术领域,具体涉及一种海洋沉积物中有机碳含量的测定方法。
背景技术:
2.海洋沉积物中的有机质含有多种物质,从简单的糖类、碳水化合物到复杂的大分子蛋白质、脂肪和有机酸等。有机碳常用于指示海洋沉积物中有机质含量,并成为海洋沉积物研究中一项十分重要的物化性质指标。有机碳含量的多少能够显著影响海洋沉积物中化合物反应途径以及海洋沉积物对大气、水等各环境介质中有机污染物的富集行为,且对于沉积物中污染物的分配和生物有效性具有重要作用。因此海洋沉积物中有机碳的测定是生态风险性评价数据库中污染分析的重要部分。
3.有机碳含量的分析可以用于定量评价分析区域的自然性质,也可以用于不同化学分析数据的标准化。现行的海洋沉积物中有机碳的分析方法分别有《海洋监测规范》(gb17378.5-2007)重铬酸钾-氧化还原容量法和热导法,以及《海洋沉积物中有机碳的测定非色散红外吸收法》(gb/t 30740-2014)。
4.《海洋监测规范》中重铬酸钾-氧化还原容量法的原理是:在浓硫酸介质中,加入一定量的标准重铬酸钾,在加热条件下将样品中的有机碳氧化成二氧化碳。剩余的重铬酸钾用硫酸亚铁标准溶液回滴,按重铬酸钾的消耗量,计算样品中有机碳的含量。
5.该方法存在如下缺陷:
6.(1)滴定终点不易确认。不同的海洋沉积物样品中均存在一定形式和含量的碳酸根离子(co
32-),如果消解完的样品中含有碳酸根离子,那么在确定滴定终点,也就是溶液的颜色由紫色突变到绿色时,随着不断摇晃溶液会很快重新呈现灰色或紫色,而继续滴定则会重新变为绿色,如此反复。这样一来,就会因个人习惯不同而造成滴定结果的误差。
7.(2)不同组分的空白样品的分析结果相差悬殊。由于不同的海洋沉积物样品中碳酸根离子的存在形式不尽相同,其分解温度和熔点也不完全一样,而海洋监测规范中没有对空白样品进行统一规定。针对此现象,李亮歌等试验了纯海砂和分别经400℃、500℃和1000℃灼烧的普通沉积物样品的相对偏差,并提出以1000℃灼烧2小时的沉积物样品作为空白样的结论。然而,不同盐分的分解温度和熔点不完全一样,温度超过600℃以上就会有板结现象发生,其原因就在于部分化合物达到了熔点。一旦灼烧温度达到熔点,就会造成板结现象,进而影响到碳酸盐的继续分解,造成滴定终点提前。尤其当灼烧温度超过800℃时,海洋沉积物中含量较高的氯化镁、氯化钠都会熔化,普通海洋沉积物空白样品会完全板结,造成测定结果明显偏高。
8.(3)没有对引发实验误差的氯离子进行有效掩蔽或处理。海洋沉积物吸附一定量的cl-,其主要影响有两个方面:一是与硫酸银反应生成氯化银(agcl)沉淀,从而降低催化效率,导致测定结果偏低;二是与重铬酸钾反应,产生氯气,导致测定结果偏高。不经掩蔽的有机碳含量随氯离子含量的增加出现了先增后降的趋势,而经过掩蔽的有机碳含量随氯离
子含量增加先保持稳定后逐渐下降。说明当氯离子含量较少时,氯离子被氧化作用占主导;随着氯离子含量增加,氯离子与银离子形成沉淀作用占主导,催化效率降低。不同的沉积物中样品氯离子含量差异较大,以滨州近岸海域海洋沉积物为例,氯离子含量最低值为1.12g/kg,最高值为4.83g/kg,近海海洋沉积物成分分析标准物质(gbw07314)中氯离子含量为4.90g/kg,而gb 17378.5-2007重铬酸钾-氧化还原容量法没有对氯离子进行有效掩蔽或处理,造成测定结果差异较大。氯离子掩蔽实验表明,加入梯度氯离子后,不经掩蔽的近海海洋沉积物成分分析标准物质(gbw07314)的有机碳含量测定值是掩蔽后的109%~122%。
9.(4)部分实验过程与步骤不合理,极易产生暴沸等现象从而导致实验失败。gb17378.5-2007重铬酸钾-氧化还原容量法采用18mm
×
160mm的试管和小漏斗,此种规格的试管一旦遇到有机碳或者氯离子含量偏高,就极易产生暴沸现象从而导致实验失败,其相邻的样品也会因受到玷污而测试失败。
10.《海洋监测规范》中热导法的原理是:样品经稀盐酸处理后,在纯氧环境中,于静态条件下燃烧960~970℃,样品中的有机碳被氧化成二氧化碳。以氦气为载气,通过仪器的热导检测器进行测定,并由测得的信号值计算有机碳的含量。
11.该方法存在如下缺陷:
12.(1)当测定钙质沉积物时,因碳酸盐含量较高,会出现正误差。通常,样品中碳酸盐(caco3)的质量分数超过10%时,正误差较为显著,尚需经过校准计算,使其结果更正确。此外,冶金、机械、原子工业及涂料、燃料、铅笔等工厂排放的沉积物中,因其含有碳(如活性碳、碳粉及石墨等),会使测定结果偏高。所以在测定上述排污口沉积物中有机碳时,应考虑其影响。
13.(2)实验过程中,因为要将样品加酸处理,经过离心机后弃去上清液,测定烘干后的样品中的有机碳含量。所以部分可溶性的有机碳会因为弃去上清液的过程而损失,从而导致实验结果偏低。
14.(3)样品需要大量步骤的预处理,繁琐费时,加之样品需要进行离心后取残渣的过程,由于样品量极少(0.1g),导致该过程极难操作。
15.(4)实验取样量太少(0.0050~0.0100g),造成误差较大。
16.《海洋沉积物中有机碳的测定非色散红外吸收法》的原理是:样品经高温燃烧或加酸处理,将相应形态的碳转化为二氧化碳,样品所产生的二氧化碳与样品中的碳含量成正比。以氧气为载体,通过红外检测器测定二氧化碳的含量即可计算相应形态碳的含量。
17.该方法存在如下缺陷:
18.(1)非色散红外吸收法采用具有非色散红外吸收(ndir)检测器的总有机碳分析仪对有机碳含量进行测定,该分析仪价格昂贵,多数海洋环境监测实验室不具备该设备,限制了该方法的推广使用。
19.(2)当样品中含有挥发性有机碳时,若采用差减法进行测定,挥发性有机碳可能在酸化过程中挥发,使测定结果产生误差。
技术实现要素:
20.针对海洋沉积物有机碳含量测定时,氯离子和碳酸盐对样品结果的干扰问题,本
发明提供一种海洋沉积物中有机碳含量的测定方法,在浓硫酸介质中,加入一定量的标准重铬酸钾,以硫酸银为催化剂,经高温消解后用分光光度计测定重铬酸钾未被还原的六价铬(cr
6+
)和被还原产生的三价铬(cr
3+
)两种铬离子的总吸光度,测试样品中的有机碳含量与总吸光度值的减少值成正比,将总吸光度值进行换算,即得测试样品中的有机碳含量。
21.本发明技术方案如下:
22.一种海洋沉积物中有机碳含量的测定方法,包括如下步骤:
23.(1)样品预处理:将测试样品、空白样品分别置于50ml聚四氟乙烯消解管中,向各聚四氟乙烯消解管中分别加入5ml纯水,摇匀,勿使结块,然后对测试样品、空白样品进行预处理,预处理方法为加入4ml磷酸溶液并摇匀,30min后加入5ml硫酸银-硫酸溶液并摇匀,放置过夜;
24.(2)建立标准曲线:将不同体积的有机碳标准贮备液置于聚四氟乙烯消解管中,分别加入纯水,使溶液体积保持5ml,摇匀,然后进行预处理,预处理方法同步骤(1);
25.预处理后,使用3ml重铬酸钾标准溶液对各溶液进行消解;消解结束后,测量铬离子的总吸光度,以零浓度校正吸光度为纵坐标,以不同体积有机碳标准贮备液对应的有机碳质量(mg)为横坐标,绘制校准曲线;
26.(3)采用重铬酸钾氧化-分光光度法对样品进行检测:按照步骤(2)方法处理预处理后的测试样品、空白样品,并记录测试样品的吸光度a和空白样品的吸光度a0;
27.(4)结果计算:将步骤(3)所得的吸光度值和步骤(2)所得的标准曲线进行比较,根据公式(ⅰ)计算得到样品中有机碳的含量,
[0028][0029]
式中,w
oc
表示测试样品干样中有机碳的含量,单位为wt%;m表示测试样品干样的取样量,单位为g;表示测试样品干样的含水率,单位为wt%;a表示标准曲线的截距;b表示标准曲线的斜率。
[0030]
进一步的,步骤(1)中,按照样品中有机碳的含量确定取样量,有机碳含量为0.00%~0.20%的,样品取样量为0.2500~0.5000g;有机碳含量为0.20%~0.50%的,样品取样量为0.1000~0.2500g;有机碳含量为0.50%~3.00%的,样品取样量为0.0300~0.100g。
[0031]
进一步的,磷酸溶液由1体积的85wt%磷酸与1体积的水混合制得;
[0032]
硫酸银-硫酸溶液的配制方法为称取10g硫酸银,加到500ml 98wt%硫酸中,搅拌1~2天,使硫酸银溶解,使用前摇匀;
[0033]
有机碳标准贮备液的配制方法为称取0.4255g邻苯二甲酸氢钾溶于适量水中,定容至1000ml,摇匀;
[0034]
重铬酸钾标准溶液的配制方法为称取9.8062g重铬酸钾溶于适量水中,定容至1000ml,摇匀。
[0035]
进一步的,步骤(2)中不同体积有机碳标准贮备液对应的有机碳质量为0~1.00mg。
[0036]
进一步的,消解方法为聚四氟乙烯消解管中加入3ml重铬酸钾标准溶液后,盖上内盖,塞紧外盖,将聚四氟乙烯消解管插入铁丝笼并置于油浴锅中,升温至195℃后恒温保温
40min,取出铁丝笼。
[0037]
进一步的,消解结束后,待消解管冷却,擦净外壁油液,用水冲洗内盖及标准溶液至50ml比色管中,加水至标线,摇匀,静置3h;取上清液至离心管中,以3500r/min离心分离10min;取上清液,用1cm比色皿,以水为参比,采用440nm
±
20nm单波长测定、600nm
±
20nm单波长测定或440nm
±
20nm与552nm双波长测定方式,测量铬离子的总吸光度,优选为采用445nm单波长进行测定。
[0038]
进一步的,步骤(4)的计算结果《0.1%时,保留到小数点后三位;计算结果≥0.1%时,保留三位有效数字。
[0039]
本发明的有益效果在于:
[0040]
碳酸盐可能影响有机碳的氧化率,本发明通过加入适量磷酸溶液摇匀后静置30min以消除干扰。磷酸是中强酸,能与无机碳快速反应。同时,由于磷酸中的磷是最高化合价,本身没有强氧化性,既不会引入新的误差因子,也不会与有机碳发生氧化还原反应,是氧化还原反应体系中最适宜与碳酸盐反应的酸。本发明采用硫酸银-硫酸溶液作为氯离子的掩蔽剂,优点一是采用液体方式,加液更准确,可以精确到0.10g硫酸银;二是加入硫酸银可以与氯离子反应生成氯化银沉淀,从而降低氯离子对实验的影响,达到掩蔽氯离子的效果;三是作为催化剂,加快氧化还原反应的进程。
[0041]
平行7次测定近海海洋沉积物成分分析标准物质(gbw07314),本方法的相对误差为0%~2.40%,相对标准偏差为2.70%~4.63%。
[0042]
当取样量为0.5g时,本方法的检出限为0.010%(以干重计),测定下限为0.040%(以干重计);当取样量为0.03g时,本方法的测定上限为3.00%(以干重计)。该检出限和测定下限分别是山东省管辖海域内海洋沉积物有机碳含量最低值的11.9%和47.6%,均远小于山东省管辖海域内海洋沉积物有机碳含量的最低值。
[0043]
在非空白样品中,随着氯离子含量从0~5mg,有机碳的测定值相对稳定,0.50mg有机碳含量样品的相对偏差为1.33%,精密度为2.71%;1.00mg有机碳含量样品的相对偏差为0.78%,精密度为0.87%;氧化率也保持相对稳定,均值分别为98.2%和90.3%。
[0044]
本方法加入4ml磷酸溶液,最大可与2.40g碳酸根离子反应,远超实际样品中碳酸根离子最大含量0.10g。
[0045]
本方法无需昂贵仪器,操作简单,适合于普通单位的推广应用。
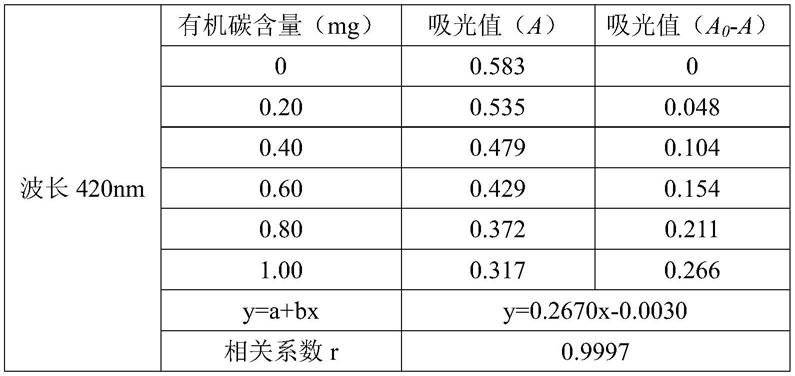
具体实施方式
[0046]
为了使本技术领域的人员更好地理解本发明中的技术方案,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
[0047]
除非另有说明,本发明具体实施方式所用试剂均为分析纯试剂,水均为新制备的超纯水或同等纯度的水。具体试剂及其配制方法如下:
[0048]
①
98wt%硫酸(h2so4):ρ=1.84g/ml,优级纯。
[0049]
②
硫酸溶液:1体积的硫酸(
①
)与9体积的水混合。
[0050]
③
85wt%磷酸(h3po4):ρ=1.69g/ml,优级纯。
[0051]
④
磷酸溶液:1体积的磷酸(
③
)和1体积的水混合。
[0052]
⑤
重铬酸钾(k2cr2o7):基准试剂或优级纯。取适量重铬酸钾研细并在120℃干燥至恒重,保存于干燥器中。
[0053]
⑥
重铬酸钾标准溶液:准确称取9.8062g重铬酸钾(
⑤
)溶于适量水中,定容至1000ml,摇匀。
[0054]
⑦
硫酸银(ag2so4)。
[0055]
⑧
硫酸银-硫酸溶液:称取10g硫酸银(
⑦
),加到500ml硫酸(
①
)中,搅拌1~2天,使之溶解,使用前小心摇匀。
[0056]
⑨
邻苯二甲酸氢钾(c8h5o4k):基准试剂或优级纯。取适量邻苯二甲酸氢钾研细并在105℃~110℃干燥至恒重,保存于干燥器中。
[0057]
⑩
有机碳标准贮备液:ρ=0.20g/l。称取0.4255g邻苯二甲酸氢钾(
⑨
)溶于适量水中,定容至1000ml,摇匀。此溶液在2~8℃下贮存,或在定容前加入约10ml硫酸溶液(
②
),常温贮存,可稳定保存一个月。
[0058]
实施例1
[0059]
一种海洋沉积物中有机碳含量的测定方法,包括如下步骤:
[0060]
(1)样品预处理:
[0061]
测试样品的采集、保存及制备按照gb 17378.1-2007、gb 17378.3-2007的规定及gb 17378.5-2007中4.1.3.2的规定执行,将制好的测试样品置于50ml聚四氟乙烯消解管中,加入5ml纯水,摇匀,勿使结块,然后加入4ml磷酸溶液并摇匀,30min后加入5ml硫酸银-硫酸溶液并摇匀,放置过夜。
[0062]
取纯海砂或普通海洋沉积物样品,500℃左右焙烧2小时后,磨细至80目作为空白样品,置于50ml聚四氟乙烯消解管中,加入5ml纯水,摇匀,勿使结块,然后加入4ml磷酸溶液并摇匀,30min后加入5ml硫酸银-硫酸溶液并摇匀,放置过夜。
[0063]
测试样品、空白样品的取样量根据下表1确定。
[0064]
表1样品中有机碳的含量与取样量的关系
[0065]
样品中有机碳的含量(%)0.00~0.200.20~0.500.50~3.00取样量(g)0.2500~0.50000.1000~0.25000.0300~0.1000
[0066]
加入磷酸后,样品会迅速产生大量气泡,说明样品中的碳酸盐快速与磷酸反应。1min后,气泡不再产生,说明碳酸盐反应完毕,10min后用ph试纸测试,ph均≤1,表明溶液中已无碳酸盐存在,为了保证充分反应,本标准将磷酸加入后等待时间定为30min。4ml磷酸溶液大约含有0.08mol氢离子,可以与0.04mol碳酸根离子反应,即最大可与2.40g碳酸根离子反应。海洋沉积物中无机碳含量大约0.50%~20.0%(近海海洋沉积物成分分析标准物质gbw07314中无机碳含量为4.70%,gbw07333中无机碳含量为1.61%,gbw07334中无机碳含量为19.9%,其中gbw07333与gbw07334中无机碳含量均按照co2含量折算)。本方法中沉积物最大取样量为0.5000g,也就是碳酸根最大含量为0.10g,4ml磷酸溶液可反应的碳酸根离子含量远超实际样品中碳酸根离子含量。
[0067]
(2)建立标准曲线:分别量取0、1.00、2.00、3.00、4.00和5.00ml有机碳标准贮备液置于50ml聚四氟乙烯消解管中,其对应的有机碳质量分别为0、0.20、0.40、0.60、0.80和1.00mg,分别加入纯水,使各聚四氟乙烯消解管内溶液体积保持5ml,摇匀,然后加入4ml磷
酸溶液并摇匀,30min后加入5ml硫酸银-硫酸溶液并摇匀,放置过夜。
[0068]
预处理后,对各溶液进行消解、冷却、定容和测试,具体方法为向聚四氟乙烯消解管中加入3ml重铬酸钾标准溶液,盖上内盖,塞紧外盖,将聚四氟乙烯消解管插入铁丝笼并置于油浴锅中,升温至195℃后恒温保温40min,取出铁丝笼;待消解管冷却后,擦净外壁的油液;用水冲洗内盖及标准溶液至50ml比色管中,加水至标线,摇匀,静置3h;取上清液至离心管中,以3500r/min离心分离10min;取上清液,用1cm比色皿,以水为参比,分别测量吸光度。
[0069]
吸收波长的选择根据待测海洋沉积物中有机碳含量确定,其中440nm
±
20nm单波长和440nm
±
20nm与552nm双波长适用于有机碳含量低于3wt%的海洋沉积物的测定,600nm
±
20nm单波长适用于有机碳含量高于1wt%、低于10wt%的海洋沉积物的测定。
[0070]
根据2019年山东省海洋环境监测数据统计,山东省管辖海域内海洋沉积物范围为0.084%~1.21%,因此440nm
±
20nm单波长和440nm
±
20nm与552nm双波长两种测定方式即可满足山东省管辖海域内海洋沉积物的检测需求;同时,考虑到双波长检测工作量为单波长检测工作量的2倍,因此,选择440nm
±
20nm单波长测定方式检测山东省管辖海域内海洋沉积物的有机碳含量。
[0071]
然后根据测量的吸光度结果,以零浓度校正吸光度为纵坐标,以不同体积有机碳标准贮备液对应的有机碳质量(mg)为横坐标,绘制校准曲线;
[0072]
(3)采用重铬酸钾氧化-分光光度法对样品进行检测:按照步骤(2)的消解、冷却、定容和测试方法处理步骤(1)得到的预处理后的测试样品、空白样品,并记录测试样品的吸光度a和空白样品的吸光度a0;
[0073]
(4)结果计算:将步骤(3)所得的吸光度值和步骤(2)所得的标准曲线进行比较,根据公式(ⅰ)计算得到样品中有机碳的含量,
[0074][0075]
式中,w
oc
表示测试样品干样中有机碳的含量,单位为wt%;
[0076]
m表示测试样品干样的取样量,单位为g;
[0077]
表示测试样品干样的含水率,单位为wt%;
[0078]
a表示标准曲线的截距;
[0079]
b表示标准曲线的斜率。
[0080]
试验例1
[0081]
通过油浴锅加热至195℃,保温10min对420nm、440nm、460nm三个波长段进行标准曲线测试,其余参数条件同实施例1步骤(2),测试数据见表2。
[0082]
表2不同波长下的标准曲线检测数据(油浴锅)
[0083][0084][0085]
可以看出440nm波长处曲线斜率最高。
[0086]
通过微波密闭消解仪进一步测试440nm、445nm、450nm三个波长的标准曲线,测试数据见表3。可以看出440nm、445nm、450nm处曲线斜率没有明显变化。
[0087]
表3不同波长下的标准曲线检测数据(微波密闭消解仪)
[0088][0089][0090]
通过油浴锅加热至195℃,保温30min分别对445nm和552nm两个波长进行测试,测试数据见表4、表5。
[0091]
表4 445nm波长下的标准曲线检测数据(油浴锅)
[0092][0093]
表5 445nm-552nm波长下的标准曲线检测数据(油浴锅)
[0094][0095][0096]
可以看出单波长和双波长测试的线性系数均大于0.9990,只是曲线斜率不同,单波长与双波长斜率比为0.959。但是,双波长测试在测试环节增加了一倍的工作量。综合考虑,优选采用单波长测试,波长选择445nm。
[0097]
试验例2
[0098]
硫酸银掩蔽有两种试剂加入顺序,一是加入重铬酸钾后再加入硫酸银-硫酸溶液(顺序a),二是加入硫酸银-硫酸溶液后再加入重铬酸钾溶液(顺序b)。
[0099]
(一)定性分析
[0100]
测试过程中发现,采取顺序a时,只要溶液中含有氯离子,溶液均呈淡黄色,说明部分氯离子被重铬酸钾氧化,没有达到完全掩蔽的效果;而采取顺序b则没有这样的现象产生。
[0101]
定性分析来看,硫酸银掩蔽宜采用顺序b,即加入硫酸银-硫酸溶液后再加入重铬酸钾溶液的顺序。
[0102]
(二)定量分析
[0103]
首先对顺序a方式预处理的样品进行氧化率测试,结果见表6。
[0104]
表6顺序a掩蔽梯度含量氯离子测试结果
[0105][0106][0107]
可以看出在空白样品(有机碳含量为0mg的样品)中,随着氯离子含量出现有机碳含量的测定值会出现0.030~0.048mg的增加,而实际空白样品中必然会有含量浓度的氯离子。将没有氯离子的样品剔除后,相对偏差为0.56%,精密度为15.72%,且主要偏差产生在氯离子含量分别为4.50mg和5.00mg两个远超海洋沉积物氯离子含量的样品中。
[0108]
在非空白样品(有机碳含量为0.50mg、1.00mg的样品)中,随着氯离子含量变化,有机碳的测定值相对稳定,0.50mg有机碳含量样品的相对偏差为0.86%,精密度为1.81%;1.00mg有机碳含量样品的相对偏差为0.45%,精密度为0.50%;氧化率也保持相对稳定,均值分别为94.5%和89.6%。
[0109]
然后对顺序b方式预处理的样品进行氧化率测试,结果见表7。
[0110]
表7顺序b掩蔽梯度含量氯离子测试结果
[0111]
[0112][0113]
可以看出在空白样品(有机碳含量为0mg的样品)中,随着氯离子含量出现有机碳含量的测定值会出现0.008~0.027mg的增加,平均值为0.019mg,远低于顺序a;相对偏差为0.61%,精密度为31.67%,没有特别明显的偏离样品。
[0114]
在非空白样品(有机碳含量为0.50mg、1.00mg的样品)中,随着氯离子含量变化,有机碳的测定值相对稳定,0.50mg有机碳含量样品的相对偏差为1.33%,精密度为2.71%;1.00mg有机碳含量样品的相对偏差为0.78%,精密度为0.87%;氧化率也保持相对稳定,均值分别为98.2%和90.3%,与顺序a相比,分别高出了3.7和0.7个百分点。
[0115]
定量分析来看,采用顺序a、顺序b均可行。
[0116]
综合比较两种试剂加入方式,采用顺序b,即加入硫酸银-硫酸溶液再加入重铬酸钾溶液的顺序能够更好地掩蔽氯离子,在同样的有机碳含量下氧化率更高,加入顺序更科学。
[0117]
试验例3
[0118]
以葡萄糖和邻苯二甲酸氢钾分别作为标准物质,测试445nm波长处标准曲线以及对近海海洋沉积物成分分析标准物质(gbw07314)的氧化率,其余条件同实施例1,结果见下表8、表9。
[0119]
表8 445nm处邻苯二甲酸氢钾标准曲线测试数据
[0120][0121]
表9邻苯二甲酸氢钾与葡萄糖氧化率对比表
[0122][0123]
可以看出,选用葡萄糖的曲线斜率较邻苯二甲酸氢钾斜率下降了2.4%;通过不同保温时间的氧化率测试,显示二者在保温40min下氧化率基本相同,在保温30min时则差异较大,葡萄糖在保温30min时明显偏低且与保温40min氧化率差异较大。
[0124]
综合比较,选用邻苯二甲酸氢钾作为有机碳测定的标准物质使用。
[0125]
试验例4
[0126]
为了测试硫酸银用量对测定结果的影响,在3份近海海洋沉积物成分分析标准物质(gbw07314)样品中各加入5ml浓度分别为10g/l、20g/l、30g/l、40g/l的硫酸银-硫酸溶液,相当于各加入0.05g、0.10g、0.15g、0.20g硫酸银,其余条件同实施例1。
[0127]
定性来看,加入0.15g和0.20g硫酸银的样品中有砖红色沉淀产生,并且加入0.20g硫酸银的样品中沉淀明显增多。说明在酸性条件下,有一部分重铬酸钾与硫酸银发生了反应,生成了铬酸银沉淀(ag2cro4)。这可能导致特征吸收波长下吸光值有所变化,从而产生误
差。因为在酸性条件下,重铬酸钾溶液一直存在如下平衡:cr2o
72-+h2o=2cro
42-+2h
+
。
[0128]
当溶液中存在大量银离子(ag
+
)的情况下,反应会向着生成铬酸银(ag2cro4)沉淀的方向进行,cro
42-+2ag
+
=ag2cro4↓
。
[0129]
定量测试结果见表10。
[0130]
表10硫酸银用量对测定结果影响
[0131][0132]
定量来看,随着硫酸银用量的增加,有机碳的测定值总体呈先增后减的趋势;加入0.10g和0.15g硫酸银的有机碳测定相对误差较小,分别为0.99%和1.99%,加入0.05g硫酸银相对误差较大,为7.15%,加入0.20g硫酸银误差最大,为15.30%,且超出标准物质浓度范围。
[0133]
综合来看,加入0.10g硫酸银用量较小,有机碳测定误差最小,最符合实验要求。
[0134]
试验例5
[0135]
以滨州近岸海域海洋沉积物为空白样品,控制取样量均为0.4900~0.5000g,测定实施例1方法的检出限和测定下限,结果见下表11。
[0136]
表11实施例1方法检出限、测定下限测试数据
[0137]
[0138]
同时委托三家验证单位使用相同空白样品及取样量,进行方法验证。三家验证单位的检出限最低为0.057%,最高为0.010%;测定下限最低为0.023%,最高为0.040%。三家验证单位检测结果得到的标准曲线还表明,有机碳含量在0~1.00mg时,可以保持良好的线性(r》0.9990),并使吸光值介于0.300~0.800之间。鉴于本方法规定称重量最小为0.0300g,故有机碳含量的最大测定值为3.00%。
[0139]
综合来看,当取样量为0.5g时,本方法的检出限为0.010%(以干重计),测定下限为0.040%(以干重计),测定上限为3.00%(以干重计)。
[0140]
尽管通过优选实施例的方式对本发明进行了详细描述,但本发明并不限于此。在不脱离本发明的精神和实质的前提下,本领域普通技术人员可以对本发明的实施例进行各种等效的修改或替换,而这些修改或替换都应在本发明的涵盖范围内/任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1