一种采用两相亲和预浓缩法对丹参中酚类化合物同时在线富集、分离方法
1.本发明属于天然药物提取检测领域,涉及一种采用两相亲和预浓缩法对丹参中酚类化合物同时在线富集、分离方法,是一种用胶束电动色谱技术在线富集酚类化合物的方法。
背景技术:
2.毛细管电泳(ce)是弥补高效液相色谱(hplc)和气相色谱(gc)不足的新兴分析技术,具有高分辨率和灵活性,应用领域广泛。目前,已经发展了各种分离模式,其中,胶束电动色谱(mekc)是重要的模式之一。1984年,由terabe等人通过在背景溶液(bgs)中加入十二烷基硫酸钠(sds),发明了mekc。随着后续发展,这一技术在食品、环境、药物、药用植物等分离科学领域得到了广泛的应用。mekc是一种优秀的混合技术,结合了色谱和电泳的分离原理,采用由表面活性剂浓度高于临界胶束浓度(cmc)的胶束假固定相,在胶束和周围水相之间分配中性和带电物质。事实上,虽然mekc丰富了ce的应用,但仍存在光路短、注入体积小、紫外灵敏度低等缺点。为了克服这些限制,离线或在线预浓缩方法已被证明是提高mekc的灵敏度的非常有效的方法。分散式液-液微萃取(dllme)-mekc,阳离子选择性耗尽进样技术(csei)-sweeping-mekc,asei-sweeping-mekc,现场增强样品注射(fesi)-mekc是在mekc中富集样品的方法。然而,开发一种新的在线预浓缩方法,用于提高复杂基质中分析物灵敏度分析仍然是有意义的。
3.近年来,基于两相物质亲和性的富集方法不断发展,其中joselito p.quirino在将表面活性剂应用于预浓缩技术以提高ce检测灵敏度方面做出了突出的贡献。2017年,joselito p.quirino等人提出了一种基于ce中分析物有效电泳速度反转的堆叠方法,称为胶束到环糊精(cd)堆积(mcds),它在动态堆叠边界带电胶束和中性环糊精之间形成。2019年,新的样品浓缩技术mcds在mekc中成功联用,用于聚焦中性、阳离子和手性分析物。2021年,他们提出了另一种基于sds和十六烷基三甲溴化铵(ctab)亲和力的富集技术,以提高紫外检测对毛细管区电泳(cze)较差的检测灵敏度。也有关于使用两相亲和力富集分析物来提高灵敏度的报道,两者都具有显著的富集效应。在此基础上,开发一种基于胶束与亚胶束亲和度的有效浓缩方法是非常必要的。
4.传统中药中所说的丹参是丹参(唇形科)的根或根茎,是一种多年生草本植物。丹参已有2000年的历史,最早记载在《神农本草经》中,属于最高级。由于丹参在防治冠心病、动脉硬化和心肌梗死方面具有副作用小和整体护理的优势,它已在世界范围内种植,并在美国和欧洲药典中均有记录。丹参的两种主要活性成分是水溶性酚酸和亲脂性丹参酮化合物,前者具有良好的生物活性,如抗氧化、抗菌、抗炎等促健康作用。酚酸是植物的次生代谢产物,在结构上构成了大量的酚类化合物,占总酚类化合物的三分之一。目前,酚酸和酚类化合物已经通过多种不同的色谱技术,如高效薄层色谱、gc、hplc和ce。然而,gc和hplc的衍生步骤和大量的流动相溶剂在酚类化合物的检测中存在局限性。有趣的是,ce具有分析时
间短、分离效率高的固有优势,逐渐开始受到越来越多的关注。具有紫外检测的cze和mekc是由电驱动分离技术作为gc和hplc的替代技术。此外,与cze相比,mekc在分离酚酸和黄酮类方面具有很大的优势。因此,建立一种在mekc中的伪固定相与样品溶液之间的亲和富集方法具有重要意义。
5.本发明采用mekc建立基于表面活性剂的两相亲和预浓缩,富集丹酚酸b等酚类化合物,在阳离子胶束中制备样品并大剂量注射。bgs中含有有机溶剂,sds在硼砂缓冲液中形成假固定相。通过优化胶束类型和浓度、样品溶液中硼砂浓度、sds浓度、乙腈含量、bgs中硼砂浓度、注射时间等一系列重要实验参数,将最佳条件应用于丹参中酚类化合物的检测。
技术实现要素:
6.本发明的目的在于针对现有技术的不足,提出一种采用两相亲和预浓缩法对丹参中酚类化合物同时在线富集、分离方法,是一种比以往富集方法更加快速高效灵敏的技术,用于具有复杂基质的样品中酚类化合物的检测。本发明利用mekc建立基于表面活性剂的两相亲和预浓缩技术检测丹参中的酚类化合物,该方法不仅实现了酚类化合物的快速检测,也极大地提高了毛细管电泳样品在线富集的灵敏度。与以往的酚类物质检测方法相比,该方法实现了其简单高效检测和分离。
7.具体地,本发明所述的方法是通过下列技术措施来实现的:
8.一种采用两相亲和预浓缩法对丹参中酚类化合物同时在线富集、分离方法包括以下步骤:
9.步骤(1)、目标分析物样品制备:
10.将丹参粉碎过筛,然后与80%(v/v)meoh混合超声30-60min,静置后取上清液;将上清液与硼砂、水混合高速离心后取上清液,得到目标分析物;
11.作为优选,硼砂浓度为20mm;
12.步骤(2)、目标分析物样品富集、分离:
13.2-1、活化毛细管柱
14.作为优选,首次使用时,毛细管柱先用1.0m naoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟;
15.2-2、对活化毛细管柱进样前进行背景溶液冲洗5-10分钟;
16.所述背景溶液包含硼砂缓冲液、乙腈和阴离子表面活性剂;
17.作为优选,所述背景溶液中硼砂浓度为0-30mm,优选为20mm;
18.作为优选,所述阴离子表面活性剂采用十二烷基硫酸钠sds;
19.作为优选,所述sds浓度为5-20mm,更为优选为10mm;
20.作为优选,所述乙腈为5-15%(v/v),更为优选为10%(v/v);
21.2-3在毛细管柱中,将样品溶液在压力进样下注射50-170s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离;其中样品溶液为目标分析物、阳离子表面活性剂、硼砂的混合液;
22.作为优选,所述样品溶液中硼砂浓度为10-30mm,优选为20mm;
23.作为优选,所述样品注射时间为140s;
24.作为优选,所述样品注射压力为50mbar;
25.作为优选,所述阳离子表面活性剂为十四烷基三甲氯化铵(ttac)、十六烷基三甲氯化铵(ctac)或十六烷基三甲溴化铵(ctab),更为优选为ctac;
26.作为优选,所述阳离子表面活性剂浓度为0.1-0.4mm,优选为0.2mm。
27.山奈酚、槲皮素、丹酚酸c和丹酚酸b的检测限(s/n=3)分别为0.0166、0.0292、0.0215和0.0195μg/ml。分析物在毛细管内迁移时间的管内精密度rsds为0.8-1.3%,峰面积的为0.4-1.8%。该方法成功地应用于丹参主要化合物酚类化合物的测定。
28.本发明的优点在于:
29.(1)利用胶束和亚胶束建立了基于表面活性剂的两相亲和预浓度。
30.(2)采用该方法进行了在mekc中酚类化合物的富集。
31.(3)将两相亲和预浓缩成功地应用于丹参中酚类化合物的检测。
32.(4)该方法简单、高效,适用于复杂基质中活性成分的检测。
附图说明
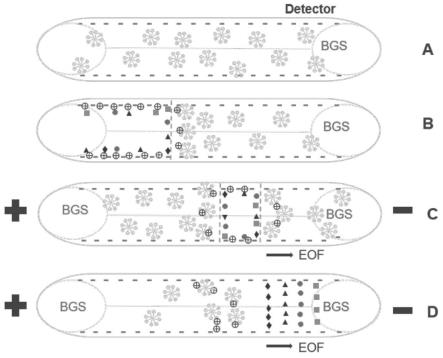
33.图1.本发明的富集机理图。
34.图2.阳离子表面活性剂种类的影响(dtac、ttac、ctab、ctac)。ce条件:样品溶液含20mm硼砂、0.2mm表面活性剂和5μg/ml混标,以50mbar下注射80s。背景溶液:20mm的硼砂,10%的acn和10mm的sds。1-山奈酚,2-槲皮素,3-丹酚酸c,4-丹酚酸b。
35.图3.ctac浓度的影响(0.1,0.2,0.3,0.4mm)。ce条件:与图2相同。
36.图4.样品中硼砂浓度的影响(0,10,20,30mm)。ce条件:与图2相同。
37.图5.sds浓度的影响(5,10,20mm)。ce条件:样品溶液含20mm硼砂、0.2mm ctac和5μg/ml混标,以50mbar下注射80s。背景溶液:20mm的硼砂,10%的acn和sds。
38.图6.acn含量的影响(5%,10%,15%(v/v))。ce条件:sds浓度选择10mm,其他条件与图5相同。
39.图7.背景溶液中硼砂浓度的影响(10,20,30mm)。ce条件:acn含量选择10%(v/v),其他条件与图6相同。
40.图8.样品注射时间的影响(50,80,110,140,170s)。分析物浓度为2.5mg/l,其他条件与图6相同。
41.图9.电泳图:a)丹参样品(140s);b)四种分析物的标准溶液(2.5μg/ml,140s)。1-山奈酚,2-槲皮素,3-丹酚酸c,4-丹酚酸b。
具体实施方式
42.如前所述,鉴于现有技术的不足,本案发明人经长期研究和大量实践,提出了本发明的技术方案,其主要是依据至少包括:
43.毛细管最初填充含有sds的bgs作为伪固定相(见图1a)。
44.含有分析物和低浓度ctac的样品溶液随后在长时间的压力注射下,引入毛细管中,其中分析物与阳离子表面活性剂ctac在静电引力作用下结合(见图1b)。一方面,部分ctac作为阳离子表面活性剂,在样品区与裸露的石英管壁上带负电荷的sio-离子结合形成涂层。另一方面,由于ctac和sds之间的静电相互作用,部分分析物在样品区右侧被释放,该
区域形成了第一个sds-ctac边界。
45.施加电压,在电渗流(eof)的作用下,样品溶液在毛细管中迁移,eof将bgs从阳极引入(见图1c)。此时,样品区左侧形成了两相的第二亲和(sds-ctac)边界,导致ctac和分析物从此界面释放。在毛细管内,样品溶液与bgs带之间产生了叠加边界,在电场作用下,样品区内的分析物在ce分离前缩小为一个小的堆叠带。最后,由于分析物性质不同,四种目标分析物以不同的速度通过检测窗口,从而实现高效检测(见图1d)。
46.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
47.一种采用两相亲和预浓缩法对丹参中酚类化合物同时在线富集、分离方法,其特征在于包括以下步骤:
48.步骤(1)、目标分析物样品制备:
49.将丹参粉碎过筛,然后与meoh混合超声30-60min,静置后取上清液;将上清液与硼砂、水混合高速离心后取上清液,得到目标分析物;
50.步骤(2)、目标分析物样品富集、分离:
51.2-1、活化毛细管柱
52.2-2、对活化毛细管柱进样前进行背景溶液冲洗5-10分钟;
53.所述背景溶液包含硼砂缓冲液、乙腈和阴离子表面活性剂;
54.2-3在毛细管柱中,将样品溶液在压力进样下注射50-170s,被扫描的分析物由胶束带到mss边界,以实现样品堆积、分离;其中样品溶液为目标分析物、阳离子表面活性剂、硼砂的混合液。
55.作为优选,步骤(1)中硼砂浓度为20mm。
56.作为优选,步骤2-1中,首次使用时,毛细管柱先用1.0m naoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min;非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
57.作为优选,步骤2-2中,所述背景溶液中硼砂浓度为0-30mm,乙腈体积含量为5-15%。
58.作为优选,步骤2-2中,所述背景溶液中硼砂浓度为20mm,乙腈体积含量为10%。
59.作为优选,步骤2-2中,所述背景溶液中阴离子表面活性剂采用十二烷基硫酸钠sds,浓度为5-20mm。
60.作为优选,步骤2-2中,所述背景溶液中十二烷基硫酸钠sds浓度为10mm。
61.作为优选,步骤2-3中,所述样品溶液中硼砂浓度为10-30mm,优选为20mm。
62.作为优选,步骤2-3中,所述样品注射时间为140s,注射压力为50mbar。
63.作为优选,步骤2-3样品溶液中所述阳离子表面活性剂为十四烷基三甲氯化铵ttac、十六烷基三甲氯化铵ctac或十六烷基三甲溴化铵ctab,浓度为0.1-0.4mm。
64.作为优选,步骤2-3样品溶液中所述阳离子表面活性剂为十六烷基三甲氯化铵ctac,浓度为0.2mm。
65.以下结合若干较佳实施例对本发明的技术方案作进一步的解释说明,但其中的实
验条件和设定参数不应视为对本发明基本技术方案的局限。并且本发明的保护范围不限于下述的实施例。
66.ce条件为:
67.检测波长:210nm。柱温:25℃。
68.分离电压:29kv。
69.毛细管柱:内径50μm,外径375μm,长度60cm,有效长度51.5cm。在第一次使用之前,新的毛细管柱要用1.0m naoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min,背景溶液冲洗10min。为了实现良好的重复性,两针间用0.1m naoh溶液冲洗2分钟,纯水冲洗2分钟,背景溶液冲洗5分钟。
70.数据记录:hp化学工作站(agilent)。
71.实施例1.考察阳离子表面活性剂种类对检测效果的影响
72.阳离子表面活性剂作为两相的关键部分之一,对四种目标分析物的检测有显著影响,对不同类型的阳离子表面活性剂产生不同的灵敏度。本实施例研究了ctab、ctac、dtac和ttac这四种阳离子表面活性剂。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
73.样品溶液含20mm硼砂、0.2mm阳离子表面活性剂和5μg/ml混合标准品,背景溶液bgs中含有20mm的硼砂,10%(v/v)的acn和10mm的sds。
74.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分离方法具体是:
75.1)活化毛细管柱
76.首次使用时,毛细管柱先用1.0mnaoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
77.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
78.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
79.所得结果如图2所示。首先讨论了ctab和ctac在同一长链化合物中不同原子的影响。结果表明,ctac(图2a)的检测结果优于ctab(图2b),具有更高的信号强度,山奈酚和丹酚酸c的峰更尖锐。随后,我们还对具有不同链长的阳离子表面活性剂进行了研究。当使用dtac时,丹酚酸c和丹酚酸b几乎没有被检测出,而槲皮素和山奈酚的峰很不规则(图2c)。随着链长的增加,被分析物的峰形变得尖锐而规则。综上所述,本发明选择ctac作为阳离子表面活性剂进行后续研究。
80.实施例2.考察ctac浓度对检测效果的影响
81.样品溶液中ctac的浓度是检测两相亲和预浓度的一个重要因素。研究了ctac浓度在0.1-0.4mm时的影响,其他条件与实施例1相一致。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
82.样品溶液含20mm硼砂、0.1-0.4mm阳离子表面活性剂ctac和5μg/ml混合标准品,背景溶液bgs中含有20mm的硼砂,10%(v/v)的acn和10mm的sds。
83.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分
离方法具体是:
84.1)活化毛细管柱
85.首次使用时,毛细管柱先用1.0mnaoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
86.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
87.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
88.结果如图3所示,在0.1mm ctac(图3a)处观察到宽峰,当浓度增加到0.2mm时,富集策略成功检测了四种化合物(图3b),这可能是因为低浓度的ctac尚未形成两相之间的亲和力。在0.3mm时,山奈酚的信号强度低于0.2mm ctac,当应用0.3mm ctac时,丹酚酸c的峰开始变形(图3c)。此外,在0.4mm ctac下,所有的峰都崩塌了,且检测灵敏度较差(图3d)。这可能是用0.2mm ctac得到bgs和样品溶液中sds最强的组合,分析物的结合和释放达到动态平衡。ctac浓度的增加在样品溶液中形成包合物,分析物在注射后没有完全释放。综上可知,采用0.2mm的ctac可以获得最佳的紫外响应。
89.实施例3.考察样品溶液中硼砂浓度对检测效果的影响
90.样品缓冲液和运行缓冲液的浓度会影响管内电场的分布,从而影响信号增强的结果。因此,对0、10、20、30mm样品中的四个硼砂浓度进行了研究,在本实施例中,样品溶液含0-30mm硼砂、0.2mm阳离子表面活性剂ctac和5μg/ml混合标准品,背景溶液bgs中含有20mm的硼砂,10%(v/v)的acn和10mm的sds。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
91.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分离方法具体是:
92.1)活化毛细管柱
93.首次使用时,毛细管柱先用1.0m naoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
94.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
95.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
96.如图4所示,当样品溶液不含硼砂时,分析物的峰面积高于样品溶液中含有10mm硼砂。然而不含硼砂的电泳图中,分析物的峰形坍塌,相比之下,含10mm硼砂的峰形更加尖锐。当硼砂浓度从10mm增加到30mm时,在20mm处得到最佳的波峰面积和最高高度。这可以解释为离子通过与管壁的相互作用,影响溶液的粘度和介电常数来影响电渗透作用。离子强度过高或过低,不利于提高分离效率。因此,我们选择了20mm的硼砂溶液作为最佳的样品缓冲液。
97.实施例4.考察sds浓度对检测效果的影响
98.为了检测胶束浓度对两相亲和预浓度的影响,使用sds作为伪固定相(在bgs中为5-20mm)。在本实施例中,样品溶液中含有5μg/ml混合标准品、0.2mm ctac、20mm硼砂,bgs由
20mm硼砂、10%acn和5-20mm sds制备。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
99.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分离方法具体是:
100.1)活化毛细管柱
101.首次使用时,毛细管柱先用1.0mnaoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
102.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
103.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
104.如图5电泳图所示,当sds浓度从5mm增加到10mm时,信号强度显著增加,杂质峰减少,基线更平稳。可以解释为,当使用5mm sds时,并没有形成胶束,sds在bgs中还是单一的表面活性剂,与ctac之间的亲和力较低。当表面活性剂浓度超过cmc(8.1mm)时,胶束形成,平衡了分析物与sds和ctac之间的作用力。当bgs中sds浓度达到20mm时,峰塌陷,基线无序。因为通过在bgs中应用较高浓度的sds,使得焦耳热的增加,从而导致了分析效果的减弱。
105.实施例5.考察acn含量对检测效果的影响
106.bgs中的有机溶剂在mekc中起着极其重要的作用,它影响着离子强度。用acn详细研究了有机溶剂用量(0、5%、10%、15%)的影响。在本实施例中,样品溶液中含有5μg/ml混合标准品、0.2mm ctac、20mm硼砂,bgs包括20mm硼砂、0-15%acn和10mm sds。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
107.将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
108.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分离方法具体是:
109.1)活化毛细管柱
110.首次使用时,毛细管柱先用1.0mnaoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
111.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
112.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
113.ce结果如图6所示。当bgs不含acn时,分析物的峰为梯形,槲皮素和山奈酚的峰未分离。从折线图可以看出,当bgs含有5%acn时,所有分析物的峰面积均小于10%,但当acn含量增加到15%时,山奈酚、槲皮素和丹酚酸b的峰面积下降。此外,被分析物的迁移时间也随着acn含量的增加而向后移动。其原因是有机溶剂的加入会降低离子强度,增加zeta电位,溶液粘度降低,改变管壁内表面的电荷分布,降低eof,延长峰值时间,提高分析效率。因此,10%的acn被认为是bgs中的最佳acn含量。
114.实施例6.考察背景溶液(bgs)中硼砂浓度对检测效果的影响
115.bgs中的硼砂浓度是影响被分析物离子强度和迁移时间的一个极其重要的因素。本实施例检测并讨论了三种硼砂浓度,即10、20和30mm。在本实施例中,样品溶液中含有5μg/ml混合标准品、0.2mm ctac、20mm硼砂,bgs包括10-30mm硼砂、10%acn和10mm sds。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
116.将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
117.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分离方法具体是:
118.1)活化毛细管柱
119.首次使用时,毛细管柱先用1.0mnaoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水冲洗2分钟。
120.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
121.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
122.通过计算,将得到的表观迁移率(μ
app
)制作成柱状图,如图7所示。当硼砂浓度从10mm增加到30mm时,四种分析物的μ
app
依次下降,说明随着bgs中硼砂浓度的增加,各分析物的迁移时间的增加而延迟。这可以解释为缓冲盐的浓度直接影响电泳介质的离子强度,从而影响zeta电位,而zeta电位的变化会影响电渗透流。随着缓冲液浓度的增加,离子强度增加,电双层厚度减小,zeta电位减小,电渗透流减小,样品在毛细管中的停留时间延长,有利于迁移时间短的组分分离和分析效率的提高。此外,在三种浓度中,20mm硼砂可以获得最好的分析效果。当硼砂浓度为10mm时,其峰面积和信号强度均小于20mm。当浓度为30mm时,峰面积减小,峰形状变宽,基线无序。同时,当硼砂浓度从10mm增加到30mm时,峰面积从19增加到45μa。这可以解释为,随着电解质浓度的增加,电解质的电导率将远远高于样品溶液,导致样品对毛细管柱的积累效应,增强样品的富集现象,增加样品的容量,从而提高分析灵敏度。然而,如果电解质浓度过高,电流增加,由于热效应引起的样品组分的峰状膨胀会使分离效应恶化。综上所述,选择bgs中含20mm硼砂作为后续实验的最佳条件。
123.实施例7.考察样品注射时间对检测效果的影响
124.样品注入时间作为预浓缩的关键环节,决定了该方法的最终富集效果。本实施例探索了50-170s的5次注射次数。。在本实施例中,样品溶液中含有2.5μg/ml混合标准品、0.2mm ctac、20mm硼砂,bgs包括20mm硼砂、10%acn和10mm sds。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。将槲皮素、山奈酚、丹酚酸b和丹酚酸c按照摩尔比1:1:1:1溶于甲醇中制备成分析用的混合标准品。
125.对丹参中4种酚类化合物(槲皮素、山奈酚、丹酚酸b和丹酚酸c)同时在线富集、分离方法具体是:
126.1)活化毛细管柱
127.首次使用时,毛细管柱先用1.0m naoh溶液冲洗25分钟,0.1m naoh溶液冲洗15min,蒸馏水冲洗10min。非首次使用时,毛细管柱先用0.1m naoh溶液冲洗2分钟,蒸馏水
冲洗2分钟。
128.2)对活化毛细管柱进样前进行背景溶液冲洗5分钟;
129.3)将毛细管柱中,将样品溶液在50mbar压力进样下注射80s,被引入的分析物由胶束带到mss边界,以实现样品堆积、分离。
130.图8记录四种化合物的峰面积并绘制成直方图,在峰面积方面,所有化合物的峰面积都随着注入时间的增加而增加,特别是当时间从50s增加到140s时,由于分析物数量的增加,所有的峰面积都大大改善。但从140增至170s,所有峰面积仅略有增加,四种化合物的峰形均有变宽和变形。原因是当注入时间过长时分析物在样品区扩散,导致峰重叠,使分离效果变差。因此,140s作为最佳样品注射时间。
131.重复性考察
132.在最佳条件下,估算了基于表面活性剂结合mekc富集酸性化合物的两相两亲性预浓缩的分析图,研究了其在丹参样品中山奈酚、槲皮素、丹酚酸c和丹酚酸b分析中的适用性。表1总结了在线性范围、回归系数、决定系数(r2)、检测限(lods)和定量限(loqs)等方面的优点分析数据。在分析物0.5-10.0μg/ml的浓度范围内得到r2:0.9993
–
0.9997,建立了良好的线性关系。山奈酚、槲皮素、丹酚酸c和丹酚酸b的lods(s/n=3)分别为0.0166、0.0292、0.0215和0.0195μg/ml。loqs(s/n=10)的变化范围在0.0553~0.0973μg/ml之间。基于上述分析的毛细管内重复性rsds(迁移时间《1.3%,《峰面积1.8%),连续3次平行进样mekc分离检测了4种混合标准溶液(2.5μg/ml),评价了新型预浓缩法的重复性。分析物在毛细管内迁移时间的管内精密度rsds为0.8%~1.3%,峰面积的为0.4%~1.8%。结果表明,建立的基于表面活性剂的两相亲和预浓缩方法具有良好的重复性,可担任丹参主要成分酚酸的检测。
133.回收率实验
134.为了确定该方法在真实样品检测中的适用性,我们将该方法对丹参中提取的主要成分酚酸进行了检测。典型的注射液(3s,50mbar)无法检测到分析物。而在两相亲和预浓缩机制下,能够有效地检测了丹参中的主要成分,丹酚酸b和丹酚酸c。为了确定该方法的准确性,我们制备了空白样品,并在优化的条件下注射,并通过在提取物中添加5μg/ml的混合标准品来评估回收率。结果令人满意,如表1所示,电泳图如图9所示。采用所开发的富集法在实际样品中检测到了痕量的山奈酚和槲皮素,这可能与提取方法有关。丹参样品中丹酚酸c和丹酚酸b的含量分别为26.5μg/ml和61μg/ml。山奈酚、槲皮素、丹酚酸c、丹酚酸b的回收率分别为91.6~96.6%。
135.表1线性回归数据、lod和loq,以及样品和加标样品的分析结果
136.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1