一种用于海洋水环境生态系统中生物有机分子原卟啉
一种用于海洋水环境生态系统中生物有机分子原卟啉
ⅸ
的检测方法
技术领域
1.本发明涉及一种原卟啉
ⅸ
的检测方法,具体涉及一种用于海洋水环境生态系统中生物有机分子原卟啉
ⅸ
的检测方法。属于有机生物分子检测技术领域。
背景技术:
2.原卟啉
ⅸ
是一种内源性荧光杂环有机化合物和基本的天然有机产物,广泛存在于地球上所有需氧生物赖以生存的环境中。原卟啉
ⅸ
是一种重要的前体物质,可介导氧运输、储存、光合作用、参与甲烷代谢等多种特殊的生理功能,也是细胞色素、过氧化氢酶、过氧化物酶和硝酸还原酶等蛋白质的共有辅基。作为一种重要的天然产物,它具有生物相容性并能与生物分子相互作用,因此,它可以用来调节分子或细胞的功能。
3.目前,原卟啉
ⅸ
在生物医学领域的应用极为广泛,如光动力疗法、抗菌和抗病毒光疗、生物传感、生物成像、抗癌药物研发和干扰生化途径等,同时被广泛应用于信号指示剂等分析行业以及军工产品的研发等多个领域。
4.海洋占全球总面积的71%,拥有着丰富的生物资源,尤其是有关海洋天然产物更是有待人类去开发利用。原卟啉
ⅸ
广泛存在于动物、植物和微生物细胞中,发挥着无法替代的生理学功能。例如,原卟啉
ⅸ
是叶绿素的唯一前体物质,在atp-螯合酶的作用下,原卟啉
ⅸ
螯合镁离子后逐步形成叶绿素,而叶绿素是绿色植物进行光合作用的主要介质,其同时介导着全球能量循环、碳循环、氮循环等多种生物地球化学循环过程。此外,微生物合成分泌的细菌叶绿素也源于原卟啉
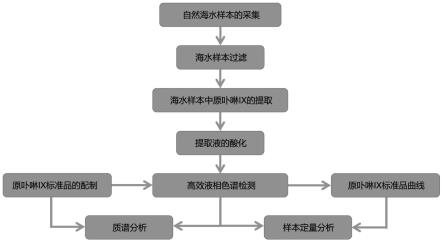
ⅸ
的合成,而微生物是地球上生物多样性的主要贡献者,在驱动碳、氮等生源要素生物地球化学循环和调节生态系统结构和功能方面发挥重要作用。因此从海洋环境中开发诸如原卟啉
ⅸ
类似的天然产物,将是今后新产品的研发与应用的关键。
技术实现要素:
5.本发明的目的是为克服上述现有技术的不足,提供一种用于海洋水环境生态系统中生物有机分子原卟啉
ⅸ
的检测方法。
6.为实现上述目的,本发明采用下述技术方案:
7.1、一种海水样本的处理方法,先将海水样本进行预过滤获得滤液,接着将滤液利用微孔滤膜进行再过滤,再过滤完成后将该微孔滤膜利用丙酮在-20℃条件下进行避光萃取20小时,得到萃取液。
8.优选的,利用孔径10μm的筛绢进行预过滤,以除去大尺寸的浮游动物、浮游植物以及碎片垃圾。
9.优选的,所述微孔滤膜为孔径0.7μm的玻璃纤维滤膜。该孔径可有效的截留海水环境中的颗粒态浮游生物,尽可能接近环境真实值。
10.优选的,微孔滤膜的直径为47mm,再过滤完成后将微孔滤膜完全浸没于5ml丙酮中
进行避光萃取。该微孔滤膜再过滤完成后会残留0.8ml水,因此萃取总体积为5.8ml,丙酮终体积浓度为86%。
11.2、上述处理方法在海洋水环境生态系统中原卟啉
ⅸ
、细菌叶绿素、叶绿素a和脱镁叶绿素定量检测中的应用。
12.3、基于前述处理方法的一种用于海洋水环境生态系统中生物有机分子原卟啉
ⅸ
的检测方法,具体步骤如下:
13.(1)先将海水样本利用前述处理方法进行处理,得到萃取液;
14.(2)调节萃取液的ph<2,室温避光酸化24小时,利用反相高效液相色谱-质谱法实现对原卟啉
ⅸ
的定量检测。
15.优选的,步骤(2)中,利用1.2mol/l盐酸溶液进行ph调节。
16.优选的,步骤(2)中,反相高效液相色谱的进样量范围为10~100μl。此范围根据样本环境变化而变化,以满足寡营养海域等特殊环境下的原卟啉
ⅸ
检测。
17.优选的,步骤(2)中,反相高效液相色谱采用的色谱柱为c18反相键合硅胶柱,柱温为34~36℃,流动相流速为1ml/min。
18.优选的,步骤(2)中,反相高效液相色谱采用荧光检测器,其激发波长为406nm,发射波长为635nm。
19.优选的,步骤(2)中,以体积百分计,反相高效液相色谱流动相组成为:流动相a:60%乙腈+39.9%水+0.1%甲酸,流动相b:99.9%丙酮+0.1%甲酸;梯度洗脱程序为:0-2分钟为20%流动相b,2-2.2分钟流动相b比例由20%梯度上升至100%,2-10分钟为100%流动相b保持,10-10.2分钟流动相b比例梯度降至20%,10.2-12分钟20%流动相b保持,整个梯度程序保持流动相比例a+b=100%。
20.优选的,步骤(2)中,质谱条件如下:离子源为电喷雾离子源,扫描模式为正离子模式,离子源温度为250℃,毛细管电压为3kv,喷嘴电压为1.5kv,雾化气压为30psi,干燥气温度为350℃,干燥气流速为11l/分钟。
21.优选的,步骤(2)中,质谱分析时,母离子为563m/z,子离子分别为504m/z,489m/z,和445m/z,对应的碰撞能分别为46ev,55ev和56ev。
22.本发明的有益效果:本发明提供了一种海水样本的处理方法,先将海水样本进行预过滤获得滤液,接着将滤液利用微孔滤膜进行再过滤,过滤完成后将该微孔滤膜利用丙酮在-20℃条件下进行避光萃取20小时,得到萃取液。本发明处理方法获得的萃取液可用于原卟啉
ⅸ
、细菌叶绿素、叶绿素a和脱镁叶绿素的定量检测。本发明所述方法及技术为国内外海洋领域首发,适用于对各类海洋环境中的原卟啉
ⅸ
进行快速、准确的定量。本发明有利于开发和利用海洋源的原卟啉
ⅸ
化合物,对海洋天然产物的开发与利用以及其在生态学上的研究具有重大意义。
23.本发明的样本来源广,提取方法简单高效,定量方法简洁快速,检测限低,回收率高,稳定性好,可在较短时间内实现样本的批量检测,检测方法的完善使得该方法不仅更适用于海洋生态系统中生物有机分子原卟啉
ⅸ
的定量检测,同时一步丙酮萃取液也可同时用于叶绿素和细菌叶绿素的定量,避免了不同处理或样本间带来的浓度差异,使之更适用于生态环境研究。本发明使用的丙酮有机试剂可极大程度地实现环境样本的细胞破碎和目标物的溶出,能较好的消除其他物质在hplc检测时带来的信号干扰,可有效保护金属化原卟
啉
ⅸ
、叶绿素和细菌叶绿素的分子结构不受破坏,较大程度的还原环境色素的真实比例。本发明所述理念及方法或可延伸至具有相同性质或特性的海洋天然产物的检测与定量,如同系卟啉化合物的检测等。
24.本发明通过高效的提取技术,极大程度的提升了原卟啉
ⅸ
的提取效果,提取方法适用于大多数自然水环境。结合高效液相色谱可实现对提取液的有效分离和定量,有利于获取、分离、回收和纯化天然原卟啉
ⅸ
化合物,高效液相色谱的利用可极大的提升检测结果的准确性和精确性,便于获得真实可靠的数据。有利于揭示原卟啉
ⅸ
在海洋环境生态系统所扮演的生态学作用,对探索全球生物地球化学循环中未知的生物化学过程具有重大研究意义。具体选择了反相高效液相色谱,采用梯度洗脱程序,可较好的实现复杂环境中原卟啉
ⅸ
分离定量。
25.本发明首次使用丙酮对自然环境中的原卟啉
ⅸ
进行提取,具体利用丙酮使细胞破碎,促使有机分子溶解或悬溶在丙酮中,后续经过酸化处理实现质子对金属化原卟啉
ⅸ
分子的中金属离子的替代,以实现环境细胞中游离和金属化原卟啉
ⅸ
的总提取,并使之呈较为单一的单分子状态,加速原卟啉
ⅸ
的溶解,该方法简单高效,短时间内可实现批量样本处理,大大的提升了检测效率,缩短了检测时间。
附图说明
26.图1为本发明实施例的海洋水环境中生物有机分子原卟啉
ⅸ
的检测流程图;
27.图2为本发明实施例中原卟啉
ⅸ
标准品的全波长扫描图;
28.图3为本发明实施例原卟啉
ⅸ
标准品的反相高效液相色谱分离图;
29.图4为本发明实施例海水样本的反相高效液相色谱分离图;
30.图5为本发明实施例参照计算的原卟啉
ⅸ
标准曲线图;
31.图6为本发明实施例海水样本中原卟啉
ⅸ
质谱鉴定图;
32.图7为本发明实施例海水样本中原卟啉
ⅸ
用不同试剂萃取效果对比图;
33.图8为本发明实施例中原卟啉
ⅸ
标准品用不同流动相分离效果色谱对比图;
34.图9为本发明实施例中原卟啉
ⅸ
标准品用不同流动相分离效果色谱峰面积对比图;
35.图10为本发明实施例海水样本中原卟啉
ⅸ
的ph范围优化色谱对比图;
36.图11为本发明实施例海水样本中原卟啉
ⅸ
的酸化时间动力学对比图;
37.图12为本发明实施例中不同地点海水样本中原卟啉
ⅸ
的浓度对比图;
38.图13为本发明实施例海水样本中原卟啉
ⅸ
与叶绿素相关性线性拟合图;
39.图14为本发明实施例海水样本中原卟啉
ⅸ
与细菌叶绿素相关性线性拟合图;
40.图15为本发明实施例海水样本中原卟啉
ⅸ
与脱镁叶绿素相关性线性拟合图。
具体实施方式
41.下面结合附图和实施例对本发明进行进一步的阐述,应该说明的是,下述说明仅是为了解释本发明,并不对其内容进行限定。
42.本发明实施例所涉及的海水样本来自于中国福建省厦门市九龙江水域和厦门湾海域,实施例样本采集时间是2021年11月。
43.实施例1:
44.一种用于海洋水环境生态系统中生物有机分子原卟啉
ⅸ
的检测方法,参考图1本发明实施例的海洋水环境中生物有机分子原卟啉
ⅸ
的检测流程图,具体步骤如下:
45.1.样本的收集与预处理
46.现场采集海水样本1000ml,用洁净的孔径为10μm的筛娟进行预过滤,除去大尺寸的浮游动物、浮游植物以及碎片垃圾,滤液储存在洁净的聚乙烯瓶中。
47.2.样本过滤与保存
48.将预过滤的样本根据水质好坏情况进行真空抽滤,过滤使用的滤膜直径为47mm,孔径为0.7μm的玻璃纤维素滤膜,记录最终过滤体积并将滤膜对折后置于5ml离心管中,于-20℃避光保存,返回实验室后应尽可能的在一个月内进行样本萃取,以防止样本因长时间放置而发生降解。
49.3.样本的萃取与酸化
50.在尽可能黑暗的条件下,将样本转移至15ml离心管中并加入5ml的纯度为100%的丙酮(47mm gf/f滤膜过滤后会残留0.8ml左右的水,因此萃取总体积为5.8ml,丙酮实际反应浓度为86%,也就是说利用终体积浓度为86%丙酮水溶液更容易实现萃取),轻微震荡混匀,确保滤膜完全浸入丙酮中,于-20℃避光萃取20小时。
51.萃取完成后将滤膜用洁净的镊子按压至管底,尽可能的转移上清至5ml的离心管中,-20℃避光保存待用。吸取500μl滤液并加入100μl去离子水配制的浓度为1.2mol/l的盐酸(即样本被稀释了1.2倍),并用0.45μm的有机系滤膜和1ml的无菌注射器进行按压过滤,混合均匀后于室温避光酸化24小时后用于原卟啉
ⅸ
检测。经0.45μm滤膜过滤的丙酮萃取液可直接用于细菌叶绿素检测。未处理的丙酮萃取液可直接用于叶绿素a和脱镁叶绿素(加1~2滴盐酸酸化30s左右)的荧光定量检测。
52.4.反相高效液相色谱
53.将萃取得到的含原卟啉
ⅸ
的萃取液通过反相高效液相色谱进行分离鉴定,具体操作条件如下:
54.高效液相色谱柱类型:c18反相键合硅胶柱,色谱柱ph耐受范围为1-10(岛津inertsustain c18,4.6
×
150mm,5μm);
55.流动相组成:流动相a:60%乙腈+39.9%水+0.1%甲酸,流动相b:99.9%丙酮+0.1%甲酸;
56.梯度洗脱程序:0-2分钟为20%流动相b,2-2.2分钟流动相b比例由20%梯度上升至100%,2-10分钟为100%流动相b保持,10-10.2分钟流动相b比例梯度降至20%,10.2-12分钟20%流动相b保持,整个梯度程序保持流动相比例a+b=100%,系统程序总计运行时间为12分钟。
57.色谱柱柱温为35
±
1℃,液相自动进样盘温度为4
±
2℃,进样体积为20~100μl,自动进样时需关闭进样器配制的照明灯,以防止样本因灯热而挥发。
58.色谱检测时荧光检测器激发波长为406nm,发射波长为635nm。
59.5.液相色谱-质谱联用鉴定海水样本中的原卟啉
ⅸ
化合物
60.质谱仪:配有agilent 1290infinity lc系统和agilent masshunter数据处理软件的agilent 6490三重串联四级杆液质联用仪;
61.色谱柱:c18反相键合硅胶柱,色谱柱ph耐受范围为1-10(岛津inertsustain c18,4.6
×
150mm,5μm)
62.流动相组成:流动相a:60%乙腈+39.9%水+0.1%甲酸,流动相b:99.9%丙酮+0.1%甲酸;
63.梯度洗脱程序:0-2分钟为20%流动相b,2-2.2分钟流动相b比例由20%梯度上升至100%,2-10分钟为100%流动相b保持,10-10.2分钟流动相b比例梯度降至20%,10.2-12分钟20%流动相b保持,整个梯度程序保持流动相比例a+b=100%,系统程序总计运行时间为12分钟。
64.色谱柱柱温为35
±
1℃,液相自动进样盘温度为4
±
2℃,进样体积为10μl,流速0.7ml/分钟。
65.离子源:电喷雾离子源
66.扫描模式:正离子模式
67.毛细管电压:3kv
68.离子源温度:250℃
69.喷嘴电压:1.5kv
70.雾化器压:30psi
71.干燥气温度:350℃
72.干燥气流速:11l/分钟
73.扫描范围:480-620m/z
74.采用上述设置条件,通过lc-ms/ms质谱扫描对化合物进行鉴定,包括以下内容:
75.根据原卟啉
ⅸ
的物质分子量(562.6g/mol)在正离子模式下扫描确定原卟啉
ⅸ
化合物的母离子为563m/z,一级子离子为504m/z,最优碰撞能为46ev;二级子离子为489m/z,最优碰撞能为55ev;三级子离子为445m/z,最优碰撞能为56。
76.根据色谱图确定目标物原卟啉
ⅸ
的色谱峰保留时间,以原卟啉
ⅸ
标准品浓度为横坐标,标准品色谱峰面积为纵坐标进行标准曲线绘制并得到相应的线性回归方程。根据样本检测所得峰面积和标准曲线进行海水样本原卟啉
ⅸ
的定量分析。
77.图2是本发明的一个实施例中原卟啉
ⅸ
标准品的全波长扫描图,以406nm波长为激发光可有效地对海水样本中的原卟啉
ⅸ
进行荧光激发。
78.图3为本发明实施例中原卟啉
ⅸ
标准品的反相高效液相色谱分离图,在所设置的液相色谱条件下,可实现原卟啉
ⅸ
标准品和叶绿素标准品的有效分离。
79.图4为本发明实施例海水样本的反相高效液相色谱分离图,参照原卟啉
ⅸ
标准品保留时间,海水样本亦可实现与其他化合物的较好的分离效果,样本保留时间范围为4.5至5.2分钟。
80.图5为本发明实施例参照计算的原卟啉
ⅸ
标准曲线图,横坐标为原卟啉
ⅸ
标准品浓度,纵坐标为响应峰面积,线性回归方程为y=2995.8869x+79.6585,其中r2=0.9989,可有效地应用于海水样本中原卟啉
ⅸ
的定量检测。
81.图6分别为本发明实施例海水样本中原卟啉
ⅸ
的液质联用质谱鉴定图,可直接说明提取物中含有目标有机分子原卟啉
ⅸ
,结合液相色谱和质谱分析结果,可定性地确认目标峰即为原卟啉
ⅸ
化合物。
82.实施例2:
83.参考图1本发明实施例的海洋水环境中生物有机分子原卟啉
ⅸ
的检测流程图,对萃取的海水样本进行如下条件优化:
84.1.萃取试剂优化
85.以甲醇、乙醇、乙腈、丙酮为萃取剂,在相同条件下分别对海水样本进行黑暗冷萃取,以丙酮/乙腈为流动相,分别对个萃取液进行酸化与未酸化色谱检测。结果如图7所示,酸化的丙酮对原卟啉
ⅸ
的萃取效果明显优于甲醇、乙醇和乙腈,且酸化要比未酸化的萃取效率更明显。国内外研究报道显示,原卟啉
ⅸ
是极微溶于水,可溶于大多数有机溶剂和酸性或碱性水溶液中。尽管甲醇也可实现卟啉类化合物的萃取,但有悖于本发明的初衷,甲醇在萃取叶绿素时会引起叶绿素的异构化和反式酯化作用,导致叶绿素分子结构发生变化,且萃取效率低于100%的丙酮,因此也不适用于本发明。本发明使用丙酮的另一目的是通过一步丙酮萃取法完成多个目标色素样本的提取,包括叶绿素、细菌叶绿素和脱镁叶绿素,真正的实现“一步多用”的目的,“一步”指一个样本只需用丙酮进行一次萃取处理,“多用”指一步丙酮萃取的溶液可同时用于原卟啉
ⅸ
、叶绿素a、脱镁叶绿素和细菌叶绿素a的检测。“一步多用”的方法在保护色素分子原有结构不受破坏的前提下,可极大程度的还原真实环境下各种色素之间的浓度分布比例,避免了不同处理或样本间带来的浓度差异,使之更适用于生态环境研究。后续原卟啉
ⅸ
的酸化步骤可实现金属化原卟啉
ⅸ
的质子替代过程,尽可能的实现环境细胞中游离的和金属化的原卟啉
ⅸ
的提取,获得的总原卟啉
ⅸ
更能体现出与其他生物色素之间的相关性,这对原卟啉
ⅸ
在生态系统中的作用研究具有极其重要的生态学和统计学意义。因此,尽管丙酮+盐酸作为萃取剂是萃取效果更好,但也不适用于本发明的“一步多用”理念,且与叶绿素等色素分子进行比较时会带来样本间误差,不适用于环境生态学研究。
86.2.流动相优化
87.将甲酸配制的原卟啉
ⅸ
标准品分别以甲醇/甲酸、乙醇/甲酸、乙腈/甲酸、丙酮/甲酸、丙酮和丙酮/乙腈/甲酸为流动相进行hplc检测,以优化不同有机溶剂对原卟啉
ⅸ
的分离效果。结果如图8和图9所示,各流动相色谱峰高由大到小顺序为丙酮/乙腈/甲酸》丙酮/甲酸》丙酮》乙醇/甲酸》甲醇/甲酸》乙腈/甲酸,且就分离度而言,以丙酮/乙腈/甲酸为流动相的色谱峰平滑效果明显好于其他流动相。色谱峰面积结果显示,含有丙酮/甲酸的流动相峰面积明显高于其他三项有机溶剂,其中乙腈/甲酸对原卟啉
ⅸ
的分离效果最弱。因此选择用高洗脱能力的丙酮/甲酸和低效率的乙腈/甲酸为流动相不仅可以确保目标物尽可能高效的被丙酮分离,也可确保非目标物被乙腈分离,这种高低洗脱相的结合更适用于本发明。
88.3.样本ph范围优化
89.以盐酸为酸度调节剂,以氢氧化钠为碱度调节剂,分别取500μl的海水样本提取液加入6个1.5ml的棕色进样瓶中,以盐酸分别调节ph值为1,3,5;以氢氧化钠调节ph值为9和11左右,以不加任何酸碱的样本作为中性样本(ph=7),混合均匀后避光反应24小时。
90.图10为本发明实施例海水样本中原卟啉
ⅸ
的ph范围优化色谱对比图,根据液相色谱检测结果可以清晰看到ph对海水样本中的原卟啉
ⅸ
的分离效果有很大的影响,在ph=1时目标物呈现较好的分离度,而在ph》3时样本呈多峰或杂峰状态,无法完成有效的分离定量。本结果亦可佐证酸性有机溶剂有利于原卟啉
ⅸ
单分子的溶解与色谱分离。
91.4.样本酸化时间优化
92.样本ph范围优化结果显示,ph对分离度有很大的影响,因此基于低酸性条件下,我们对样本的酸化时间进行了动力学监测。将高效液相色谱仪自动进样盘温度设置为室温,以1小时为间隔对同一酸化的样本进行连续24小时的监测,每组样本设置5和平行样。监测结果以酸化时间为横坐标,样本浓度变化为纵坐标绘制趋势图。
93.结果如图11所示,图11为本发明实施例海水样本中原卟啉
ⅸ
的酸化时间动力学对比图,可看出随着酸化时间的不断延长,样本酸化产出的原卟啉
ⅸ
浓度也在逐渐的上升,图11插图表示酸化时间分别在0小时、12小时和24小时的色谱图,可以清楚的看到随着酸化时间的延长,色谱峰发生了明显的变化。不断上升的原卟啉
ⅸ
浓度可能是因为氢离子替代金属离子的速度不同所致。对比最后3小时的原卟啉
ⅸ
浓度变化,结果显示3者浓度并无显著性差异,因此,基于时效性及实际应用的考虑,建议酸化时间统一确立为24小时。
94.实施例3:
95.根据实施例1和实施例2检测方法,对本发明所建立的方法进行方法学验证,具体内容如下:
96.1.检测限和定量限
97.将0.2nmol/l的原卟啉
ⅸ
标准品注入液相色谱中以进行检测限和定量限的确定,含6次平行样。将原卟啉
ⅸ
的平均峰面积定义为响应信号,以6次平行样峰面积的相对标准差为背景噪音计算信噪比,检测限定义为3倍信噪比所对应的原卟啉
ⅸ
浓度,定义10倍信噪比所对应的原卟啉
ⅸ
浓度为定量限。
98.检测结果显示信噪比为3.8,检测限为3.83
±
1.00pm,定量限为12.77
±
3.34pm,说明本发明所述方法具有极低的检测限和定量限,可适用于环境含量较低的水环境中原卟啉
ⅸ
的检测与定量。
99.2.准确度、精确性和回收率验证
100.为了验证同一天内不同批次间以及不同天内样本检测的准确性和精确性,我们选择了4个浓度梯度的原卟啉
ⅸ
进行了验证。原卟啉
ⅸ
浓度梯度分别设置为0.5nm,1nm,5nm和60nm。天内的检测间隔时常为6小时,天外间隔是4天。选用标准品浓度为1nm/ml的原卟啉
ⅸ
进行回收率测定,标准品按照实施例1所述步骤进行回收,按照实施例2所述步骤进行回收率验证,以检测值和真实值比例表示回收率。
101.结果如表1所示,天内不同浓度水平的平均准确度在80~95%之间,精确性误差在0.3~14%之间;天外准确度在90~99%之间,精确性误差在2~4.6%之间,表明仪器检测相对精准。回收率平均范围为97.48
±
1.92%,回收效率较高。
102.表1.本发明实施例海水样本中原卟啉
ⅸ
的准确度和精确性验证结果
[0103][0104]
实施例4:
[0105]
根据实施例1和实施例2检测方法,对实际环境样本进行原卟啉
ⅸ
定量检测。样本于2021年11月在福建省厦门市九龙江水域和厦门湾海域进行海上航次调查时所采,现场采集表层0.5-1米的海水样本以及底层海水样本各1000ml,按实施例所述方法进行样本的收集与处理。按实施例2所述方法进行海水样本中原卟啉
ⅸ
的定量和定性检测。
[0106]
检测结果如图12和表2所示,结果显示原卟啉
ⅸ
的浓度会随着海水盐度的变化而变化,具体体现在九龙江上游盐度较低的样本原卟啉浓度要高于厦门湾盐度高的样本浓度,且表层样本原卟啉
ⅸ
含量在九龙江上游要高于底层样本含量,而九龙江口则相反,厦门湾表底层含量趋于一致。
[0107]
表2.本发明实施例中九龙江-厦门湾海水样本原卟啉
ⅸ
检测结果
[0108][0109][0110]
由于原卟啉
ⅸ
是叶绿素和细菌叶绿素的直接前体物质,因此我们对比分析了相同环境下原卟啉
ⅸ
与叶绿素、细菌叶绿素以及脱镁叶绿素之间的关系。图13、图14和图15分别为本发明实施例中原卟啉
ⅸ
与叶绿素a、细菌叶绿素a及脱镁叶绿素之间相关性线性拟合
图,其与叶绿素、细菌叶绿素和脱镁叶绿素之间的表层线性拟合r2分别为0.8946、0.8476和0.9061,底层原卟啉
ⅸ
与底层叶绿素、细菌叶绿素和脱镁叶绿素的线性拟合r2分别为0.6949、0.6244和0.8108,这说明了自然海水样本中原卟啉
ⅸ
的确与其衍生物叶绿素、细菌叶绿素及脱镁叶绿素存在很大的正相关关系,为原卟啉
ⅸ
有机生物分子在生态学中的应用提供的可靠的数据和技术支撑。
[0111]
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1