芪珠升白口服液的质量评价方法
1.本发明属于药品质量控制领域,涉及一种药物质量评价方法,尤其涉及一种芪珠升白口服液的质量评价方法。
背景技术:
2.白细胞减少症通常是由感染、药物毒副作用等多种原因引起的一组综合征,临床诊断依据为外周血中每升白细胞总数持续低于4.0
×
109,是迄今为止临床内科的常见病,继发性和原因不明性为两种临床常见发病原因,而由于放射线照射的物理治疗和抗肿瘤药物的毒副作用继发于肿瘤化疗、放疗后的白细胞减少症最为常见,并会导致患者机体抵抗力能力下降,增加了感染的风险,进而危及生命;并且由于可能影响正常化疗周期的用药,而成为患者病程迟迟难以结束的主要障碍。因此,升高白细胞、改善机体功能、提高患者的抵病能力,成为肿瘤放化疗的一种重要治疗手段和方法。
3.芪珠升白口服液是在长期临床实践基础上,结合民间使用经验,研究的由区域性特色中药资源为主药组成的一种新型的用于放化疗致白细胞减少的复方中药制剂。该制剂由珠子参、黄芪、女贞子、当归、灵芝和甘草组成,提取纯化技术采用水煎煮法和离心法等,具有服用剂量少,质量稳定等特点,前期药理实验证明,该制剂具有升高白细胞、提高机体免疫能力、加强化疗药物的细胞敏感性及降低来自肿瘤放、化疗带来的不良反应等作用,而且毒副作用小。
4.芪珠升白口服液的处方配比及制备工艺是:黄芪15g,珠子参6g,女贞子9g,甘草6g,灵芝10g,当归6g,16倍量水,煎煮提取3次,每次1.5小时,滤过,合并滤液,药液60℃减压浓缩到药材的3倍量,高转速(6000~25000r/min)离心20分钟,取上清液,浓缩药液,以生药量计算为0.312g/ml,加入矫味剂为15%的蔗糖、抑菌剂为0.15%的山梨酸钠,混匀,按规格进行分装。
5.目前对芪珠升白口服液主要药效学包括升高白细胞和改善免疫功能有初步研究,未对芪珠升白口服液所含化学成分进行薄层色谱法定性鉴别及定量分析,难以准确控制药品质量。
技术实现要素:
6.为了解决背景技术中存在的上述技术问题,本发明提供了一种芪珠升白口服液的质量评价方法。
7.为了实现上述目的,本发明采用如下技术方案:
8.一种芪珠升白口服液的质量评价方法,其特征在于:所述芪珠升白口服液的质量评价方法包括以下步骤:
9.1)采用薄层色谱法对芪珠升白口服液中的原料进行定性鉴别;
10.2)根据高效液相色谱法对原料中的活性成分进行定量分析;
11.3)结合芪珠升白口服液的相对密度、ph值、步骤1)所得到的定性鉴别结果以及步
骤2)所获取得到的定量分析结果对芪珠升白口服液的质量进行评价。
12.上述步骤1)中的原料包括黄芪、珠子参以及当归。
13.上述步骤1)中的原料是黄芪时,所述步骤1)的具体实现方式是:
14.取芪珠升白口服液20ml,加水饱和正丁醇萃取2次,每次20ml,弃去水液,合并正丁醇液,再以氨试液洗涤2次,每次20ml,弃去洗液;正丁醇液水浴蒸干,残渣加甲醇4ml溶解,制成供试品溶液;根据供试品溶液的制备方法,各取阴性对5g制成阴性对照溶液,阴性对照物是珠子参、当归、女贞子、灵芝以及甘草;另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的对照品溶液;分别吸取供试品溶液、阴性对照溶液以及对照品溶液各5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105℃加热至斑点显色清晰,分别在日光和紫外光下检视;供试品色谱中,在与对照品色谱相应的位置上,日光下显相同棕褐色斑点;紫外光下显相同颜色的橙黄色荧光斑点;所述三氯甲烷-甲醇-水的体积比是15:7:2;所述紫外光的波长是365nm。
15.上述步骤1)中的原料是珠子参时,所述步骤1)的具体实现方式是:
16.取芪珠升白口服液5ml,加水饱和正丁醇萃取2次,每次10ml,弃去水液,合并正丁醇液。正丁醇液水浴蒸干,残渣加甲醇2ml溶解,制成供试品溶液;取珠子参对照药材1g制成对照药材溶液;根据供试品溶液的制备方法,各取阴性对照物5g制成阴性对照溶液,所述阴性对照物是黄芪、当归、女贞子、灵芝以及甘草;另取人参皂苷ro对照品,加甲醇制成每1ml含2mg的对照品溶液;分别吸取供试品溶液、对照药材溶液、阴性对照溶液以及对照品溶液各5μl,分别点于同一硅胶g薄层板上,以正丁醇-乙酸乙酯-甲醇-甲酸-水的上层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105℃加热至斑点显色清晰,在紫外光下检视;在与对照品色谱相应的位置上,紫外光下显相同颜色的荧光斑点;所述正丁醇-乙酸乙酯-甲醇-甲酸-水的体积比是15:10:0.5:0.3:2;所述紫外光的波长是365nm。
17.上述步骤1)中的原料是当归时,所述步骤1)的具体实现方式是:
18.取芪珠升白口服液10ml,加乙醚萃取2次,每次15ml,弃去水液,合并乙醚液;乙醚液水浴蒸干,残渣加乙醇2ml溶解,制成供试品溶液;取当归对照药材1g制成对照药材溶液;根据供试品溶液的制备方法,各取阴性对照物5g制成阴性对照溶液,所述阴性对照物是黄芪、珠子参、女贞子、灵芝以及甘草;吸取供试品溶液、对照药材溶液以及阴性对照溶液各10μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯为展开剂,展开,取出,晾干,在紫外光下检视;供试品色谱中,在与对照品色谱相应的位置上,紫外光下显相同颜色的荧光斑点且阴性无干扰;所述正己烷-乙酸乙酯的体积比是2:1;所述紫外光的波长是365nm。
19.上述步骤2)是根据高效液相色谱法对原料中女贞子的活性成分、珠子参的活性成分以及甘草的活性成分进行定量分析。
20.上述步骤2)是根据高效液相色谱法对原料中女贞子的活性成分特女贞苷、珠子参的活性成分人参皂苷ro和竹节参皂苷ⅳa以及甘草的活性成分甘草酸铵进行定量分析。
21.上述定量分析的具体实现方式是:
22.2.1)准备混合对照品溶液、供试品溶液以及阴性样品溶液;
23.2.2)采用相同的色谱条件对步骤2.1)所准备的混合对照品溶液、供试品溶液以及阴性样品溶液进行定量分析。
24.上述步骤2.1)中混合对照品溶液的具体配制方式是:精密称取特女贞苷17.08mg、人参皂苷ro 10.00mg、竹节参皂苷ⅳa 8.56mg以及甘草酸铵对照品8.56mg,置同一棕色容量瓶中,向容量瓶中加甲醇10ml,制成含特女贞苷1.708mg
·
ml-1
、人参皂苷ro 1.00mg
·
ml-1
、竹节参皂苷ⅳa 0.856mg
·
ml-1
以及甘草酸铵0.856mg
·
ml-1
的混合对照品溶液;
25.所述供试品溶液的具体配制方式是:取芪珠升白口服液2ml,称取d101大孔树脂5g,通过d101大孔树脂柱,依次用2bv水,3bv 20%乙醇,5bv 60%乙醇洗脱,收集60%乙醇洗脱液,蒸干,残渣用甲醇溶解于5ml量瓶中,用甲醇稀释至刻度,摇匀,作为供试品溶液;
26.所述阴性样品溶液包括不含女贞子的芪珠升白口服液阴性样品溶液、不含珠子参的芪珠升白口服液阴性样品溶液以及不含甘草的芪珠升白口服液阴性样品溶液;
27.所述不含女贞子的芪珠升白口服液阴性样品溶液的具体配制方式是:根据常规工艺,配置不含女贞子的芪珠升白口服液2ml,称取d101大孔树脂5g,通过d101大孔树脂柱,依次用2bv水,3bv 20%乙醇,5bv 60%乙醇洗脱,收集60%乙醇洗脱液,蒸干,残渣用甲醇溶解于5ml量瓶中,用甲醇稀释至刻度,摇匀,作为不含女贞子的芪珠升白口服液阴性样品溶液;
28.所述不含珠子参的芪珠升白口服液阴性样品溶液的具体配制方式是:根据常规工艺,配置不含珠子参的芪珠升白口服液2ml,称取d101大孔树脂5g,通过d101大孔树脂柱,依次用2bv水,3bv 20%乙醇,5bv 60%乙醇洗脱,收集60%乙醇洗脱液,蒸干,残渣用甲醇溶解于5ml量瓶中,用甲醇稀释至刻度,摇匀,作为不含珠子参的芪珠升白口服液阴性样品溶液;
29.所述不含甘草的芪珠升白口服液阴性样品溶液的具体配制方式是:根据常规工艺,配置不含甘草的芪珠升白口服液2ml,称取d101大孔树脂5g,通过d101大孔树脂柱,依次用2bv水,3bv 20%乙醇,5bv 60%乙醇洗脱,收集60%乙醇洗脱液,蒸干,残渣用甲醇溶解于5ml量瓶中,用甲醇稀释至刻度,摇匀,作为不含甘草的芪珠升白口服液阴性样品溶液。
30.上述步骤2.2)中色谱条件是:色谱柱:inertsil ods-3色谱柱,所述色谱柱的规格是4.6mm
×
250mm,色谱柱的粒径是5μm;流动相乙腈(a)-0.1%磷酸水(b),洗脱程序是0~25min,15%~20%a;25~37min,20%~25%a;37~52min,25%~30%a;52~55min,30%~35%a,55~60min,35%~40%a;60~70min,40%~42%a;检测波长203nm;流速1.0ml
·
min-1
;进样量10μl;柱温30℃。
31.本发明的优点是:
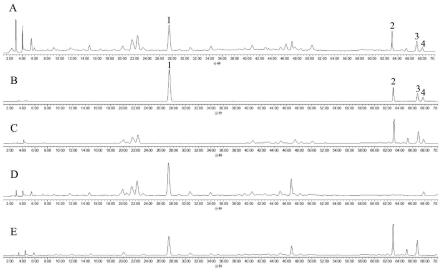
32.本发明提供了一种芪珠升白口服液的质量评价方法,进而为质量标准提供依据。本发明采用薄层色谱(tlc)法对黄芪、珠子参和当归进行定性鉴别,采用waters e2695型高效液相色谱仪,选用inertsil ods-3色谱柱,以乙腈-0.1%磷酸水为流动相梯度洗脱,检测波长为203nm测定方中主要药效成分特女贞苷、人参皂苷ro、竹节参皂苷iva、甘草酸铵含量。结果:tlc斑点分离度好,阴性无干扰;特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵质量浓度分别在0.1708~1.708mg
·
ml-1
、0.1~1.0mg
·
ml-1
、0.0856~0.856mg
·
ml-1
和0.0856~0.856mg
·
ml-1
范围内线性关系良好,平均加样回收率分别为98.26%、95.90%、95.76%、98.09%,rsd分别为1.68%、2.78%、1.84%、2.58%。该方法稳定可行,可用于芪珠升白口服液质量评价。本发明建立的芪珠升白口服液的质量评价方法可用于芪珠升白口服液的质量评价与控制,也可为含有本方药材的复方制剂含量测定提取方法提供参考。
附图说明
33.图1是hplc色谱图;
34.图2是黄芪的tlc色谱图;
35.图3是珠子参的tlc色谱图;
36.图4是当归的tlc色谱图。
具体实施方式
37.芪珠升白口服液是陕西中医药大学附属医院在长期临床实践基础上,结合民间使用经验,由黄芪、女贞子、珠子参、当归、灵芝、甘草组成的一种用于放化疗致白细胞减少的复方中药制剂,该制剂具有升高白细胞、增强机体免疫功能的作用。目前对其主要药效学包括升高白细胞和改善免疫功能有初步研究,未对制剂所含化学成分进行薄层色谱法定性鉴别及定量分析,难以准确控制药品质量。本研究通过采用薄层色谱法对黄芪、珠子参、当归进行定性鉴别;建立高效液相色谱(hplc)法对女贞子中活性成分特女贞苷、珠子参中活性成分人参皂苷ro和竹节参皂苷ⅳa、甘草中活性成分甘草酸铵进行定量分析,并参照2020年版《中国药典》(四部)对芪珠升白口服液的相对密度、ph值等进行测定,以期为芪珠升白口服液的质量控制提供参考。
38.1.材料
39.1.1仪器
40.waters e2695型高效液相色谱仪(包括自动进样器,四元泵,柱温箱,2998pda检测器);sq8200hd型超声波清洗器(上海冠特超声仪器有限公司);hh-2型恒温水浴锅(北京科伟永兴仪器有限公司);gb204型电子天平(万分之一,瑞士梅特勒-托利多公司);远红外辐热炉(米技电子电器上海有限公司)。
41.1.2药品与试剂
42.黄芪、珠子参、女贞子、当归、灵芝、甘草饮片均购自陕西兴盛德药业有限责任公司,经检验均符合陕西省中药饮片标准;特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵(批号分别为hs19515s1、hs19115b2、hs19215b1、hm19115s2,宝鸡辰光生物科技有限公司,含量:98%);黄芪甲苷(中国食品药品检定研究院,批号:0781-20008);色谱乙腈、磷酸(天津市科密欧化学试剂有限公司,分析纯);d101大孔树脂(上海蓝季科技发展有限公司,批号:170625);硅胶g板(青岛海洋化工有限公司,批号:20190903)。
43.2方法与结果
44.2.1含量测定
45.2.1.1色谱条件
46.色谱柱:inertsil ods-3色谱柱(4.6mm
×
250mm,5μm);流动相乙腈(a)-0.1%磷酸水(b),洗脱程序(0~25min,15%~20%a;25~37min,20%~25%a;37~52min,25%~30%a;52~55min,30%~35%a,55~60min,35%~40%a;60~70min,40%~42%a);检测波长203nm;流速1.0ml
·
min-1
;进样量10μl;柱温30℃。
47.2.1.2混合对照品溶液
48.精密称取特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵对照品17.08mg、10.00mg、8.56mg、8.56mg,置棕色容量瓶中,加甲醇10ml,制成特女贞苷1.708mg
·
ml-1
、人参
皂苷ro 1.00mg
·
ml-1
、竹节参皂苷ⅳa 0.856mg
·
ml-1
、甘草酸铵0.856mg
·
ml-1
的混合对照品溶液。(10℃以下保存)。
49.2.1.3供试品溶液
50.取口服液2ml,称取d101大孔树脂5g,通过d101大孔树脂柱,依次用2bv水,3bv 20%乙醇,5bv 60%乙醇洗脱,收集60%乙醇洗脱液,蒸干,残渣用甲醇溶解于5ml量瓶中,用甲醇稀释至刻度,摇匀,作为供试品溶液,备用。
51.2.1.4阴性样品溶液
52.按处方比例及生产工艺分别制备不含女贞子、珠子参、甘草药材的口服液阴性样品,按2.1.3项下方法制备,即得。
53.2.1.5专属性考察
54.取供试品溶液、对照品溶液、阴性样品溶液,按2.1.1项下色谱条件进行测定,阴性无干扰。结果见图1,图1中,a是供试品溶液、b是对照品溶液、c是不含女贞子的阴性对照溶液、d是不含珠子参的阴性对照溶液、e是不含甘草的阴性对照溶液,1#峰代表特女贞苷、2#峰代表人参皂苷ro、3#峰代表竹节参皂苷ⅳa、4#峰代表甘草酸铵。
55.2.1.6线性关系考察
56.精密吸取2.1.2项下制备好的混合对照品溶液2ml、1.5ml、1ml、0.5ml、0.2ml置2ml量瓶中,加甲醇定容至刻度。在2.1.1项下条件测定峰面积,以对照品质量浓度(x)与峰面积(y)作标准曲线,得回归方程,结果见表1。
57.表1 4种指标成分的线性范围考察结果
[0058][0059]
2.1.7精密度试验
[0060]
精密吸取混合对照品溶液10μl,连续重复进样6次,按2.1.1项下色谱条件测定峰面积,结果特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵峰面积的rsd分别为0.41%、0.58%、0.98%、1.09%,表明该仪器精密度良好。
[0061]
2.1.8重复性试验
[0062]
精密移取同一批口服液样品(批号:210301)2ml,按2.1.3项下方法制备,平行6份,精密吸取10μl,注入液相色谱仪测定,特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵平均含量分别为0.704mg
·
ml-1
、0.745mg
·
ml-1
、0.595mg
·
ml-1
、0.432mg
·
ml-1
,rsd分别为1.66%、2.06%、2.02%、1.79%,表明本方法重复性良好。
[0063]
2.1.9稳定性试验
[0064]
取口服液(批号:210301)分别在0、3、6、9、12、24h按2.1.1项下色谱条件测定,结果特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵峰面积的rsd为1.44%、1.32%、1.87%、2.01%,表明本品24h内稳定性良好。
[0065]
2.1.10加样回收率试验
[0066]
取重复性试验项下的样品1ml,平行6份,精密加入特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵对照品适量,再按2.1.3项下方法制备,测定含量,平均回收率分别为98.26%、95.90%、95.76%、98.09%,rsd分别为1.68%、2.78%、1.84%、2.58%,表明回收率良好,结果见表2。
[0067]
表2回收率试验结果(n=6)
[0068][0069][0070]
2.1.11耐用性试验
[0071]
本试验考察了inertsil ods-3色谱柱(4.6mm
×
250mm,5μm)和thermobds hypersil c
18
色谱柱(5μm,4.6mm
×
150mm),各组分均能够达到较理想分离度,该方法耐用性
良好。
[0072]
2.1.12样品含有量测定
[0073]
取8批口服液,按2.1.3项下方法制备,按2.1.1项下条件测定。结果见表3。计算8批样品中特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵的平均含量分别为0.683mg
·
ml-1
、0.718mg
·
ml-1
、0.605mg
·
ml-1
、0.540mg
·
ml-1
。
[0074]
表3含有量测定结果(n=1)
[0075][0076]
2.2tlc法定性鉴别
[0077]
2.2.1黄芪
[0078]
参照2020年版《中国药典》四部通则0502薄层色谱法,取口服液20ml,阴性对照5g,制成供试品溶液、阴性对照溶液;另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的对照品溶液。吸取上述三种溶液各5μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水(15:7:2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105℃加热至斑点显色清晰,分别在日光和紫外光(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,日光下显相同棕褐色斑点;紫外光下显相同颜色的橙黄色荧光斑点。结果见图2,图2中,a是日光下黄芪tlc色谱图、b是紫外光下黄芪tlc色谱图,1~3分别是供试品溶液;4是黄芪甲苷;5是阴性对照溶液。阴性无干扰,斑点分离较好。
[0079]
2.2.2珠子参
[0080]
参照2020年版《中国药典》四部通则0502薄层色谱法,取口服液5ml,对照药材1g,阴性对照5g,制成供试品溶液、对照药材溶液、阴性对照溶液;另取人参皂苷ro对照品适量,加甲醇制成每1ml含2mg的对照品溶液。吸取上述四种溶液各5μl,分别点于同一硅胶g薄层板上,以正丁醇-乙酸乙酯-甲醇-甲酸-水(15:10:0.5:0.3:2)的上层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105℃加热至斑点显色清晰,在紫外光(365nm)下检视。结果见图3,图3中1~3.供试品溶液;4.对照药材溶液;5.人参皂苷ro;6.阴性对照溶液。在与对照品色谱相应的位置上,紫外光下显相同颜色的荧光斑点,阴性无干扰,斑点分离较好。
[0081]
2.2.3当归
[0082]
参照2020年版《中国药典》一部当归项下薄层色谱鉴别和2020年版《中国药典》四部通则0502薄层色谱法,取口服液10ml,当归对照药材1g,阴性对照5g,制成供试品溶液、对
照药材溶液、阴性对照溶液。吸取上述三种溶液各10μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯(2:1)为展开剂,展开,取出,晾干,在紫外光(365nm)下检视。结果见图4,图4中,1~3.供试品溶液;4.对照药材溶液;5.阴性对照溶液。供试品色谱中,在与对照品色谱相应的位置上,紫外光下显相同颜色的荧光斑点且阴性无干扰。
[0083]
2.3检查
[0084]
2.3.1相对密度
[0085]
参照相对密度测定法(中国药典2020版四部0601)对3批样品进行测定。结果见表4。
[0086]
表4相对密度测定结果(n=3)
[0087][0088]
2.3.2 ph值
[0089]
参照ph值测定法(中国药典2020版四部0631)对3批样品测定。结果见表5。
[0090]
表5 ph值结果(n=3)
[0091][0092]
本发明分别对芪珠升白口服液中黄芪、珠子参、当归进行薄层鉴别,黄芪参照文献与2020年版《中国药典(一部)》确定采用正丁醇萃取后氨试液洗涤的方法鉴别黄芪甲苷,所得图谱斑点清晰分离度较好。珠子参与当归处理方法同2020年版《中国药典(一部)》所收录的方法,并对当归展开条件优化为正己烷-乙酸乙酯(2:1)使其比移值适中,斑点清晰。
[0093]
芪珠升白口服液配方中女贞子、珠子参、甘草在方中作为补益药,据报道其均具有免疫调节或升高白细胞的作用,根据2020年版《中国药典》对特女贞苷、人参皂苷ro、竹节参皂苷ⅳa、甘草酸铵进行定量分析,分别考察不同流动相甲醇-水,乙腈-水、乙腈-0.1%甲酸水与乙腈-0.1%磷酸水,最终选取乙腈-0.1%磷酸水梯度洗脱后图谱基线平稳,分离度较好。本方为水煎液并含6味药材,成分复杂且极性偏大,分别采用50%甲醇和稀乙醇稀释、正丁醇萃取、大孔树脂洗脱收集的样品处理方法,结果表明直接稀释与液液萃取的方法虽简单,可操作性好但样品杂质过多影响较大,目标成分与杂质峰较难达到有效分离,根据文献与目标成分特点选择d101大孔树脂进行提取,经预实验确定以2bv水,3bv 20%乙醇,5bv 60%乙醇2bv
·
h-1
洗脱后收集60%乙醇洗脱液,此时供试品溶液提取充分且无干扰,重复性,稳定性与回收率均良好。本发明建立的薄层鉴别和含量测定方法可用于芪珠升白口服液的质量评价与控制,也可为含有本方药材的复方制剂含量测定提取方法提供参考。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1