一种采用一测多评法测定茯苓中茯苓酸A和茯苓酸B含量的方法与流程
37%;35-36min,流动相a为63%-55%,流动相b为37%-45%;36-45min,流动相a为55%,流动相b为45%。
15.本技术的有益效果包括但不限于:
16.1.根据本技术的采用一测多评法测定茯苓中茯苓酸a和茯苓酸b含量的方法,该方法可为检测茯苓配方颗粒所用茯苓药材和饮片提供参考,本方法两个成分理论塔板数均大于10000,成分保留及洗脱能力佳,方便快捷成本低,特异性强、灵敏度高,便于普及应用。
17.2.根据本技术的采用一测多评法测定茯苓中茯苓酸a和茯苓酸b含量的方法,通过限定提取溶剂的体积分数、提取方式和时间及称样量的克数,保证溶液中茯苓酸a和茯苓酸b的含量较高,检测效果好。
18.3.根据本技术的采用一测多评法测定茯苓中茯苓酸a和茯苓酸b含量的方法,该方法首次通过高效液相一测多评技术实现了茯苓药材和饮片中三萜酸类成分茯苓酸a与茯苓酸b的定量,为《中国药典》和行业监管提供参考。
19.4.根据本技术的采用一测多评法测定茯苓中茯苓酸a和茯苓酸b含量的方法,该方法采用一个波长下同时检测茯苓酸a和茯苓酸b两种成分,信号相应强,不需要切换检测波长,检验效率高,成本低,适用性强。
附图说明
20.此处所说明的附图用来提供对本技术的进一步理解,构成本技术的一部分,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。在附图中:
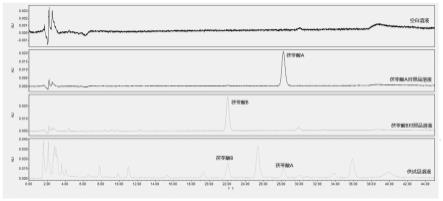
21.图1为本技术实施例涉及的茯苓丁专属性图谱;
22.图2为本技术实施例涉及的茯苓酸b标准曲线;
23.图3为本技术实施例涉及的茯苓酸a标准曲线。
具体实施方式
24.下面结合实施例详述本技术,但本技术并不局限于这些实施例。
25.除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。本发明所使用的试剂或原料均可通过常规途径购买获得,如无特殊说明,本发明所使用的试剂或原料均按照本领域常规方式使用或者按照产品说明书使用。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。本专利中所述的较佳实施方法与材料仅作示范之用。
26.其中,本技术中涉及甲醇的百分比,若无特殊说明,均为体积分数。
27.实施例1
28.1.仪器与试药
29.仪器:waters高效液相色谱仪,agilent高效液相色谱仪,bsa224s-cw电子天平(赛多利斯科技仪器(北京)有限公司),ps-60al超声波清洗器(山东东岳国际经贸合作股份有限公司),kdm型可调控温电热套(山东鄄城华鲁电热仪器有限公司)
30.色谱柱:luna c18sn:h20-403862(4.6*250mm,5μm);
31.luna c18sn:h21-337252(4.6*250mm,5μm)。
32.试药:
33.茯苓丁(批号:2201001,厂家:亳州市圣海中药饮片有限公司;)
34.茯苓酸b对照品(批号:9521,上海诗丹德,纯度:98.0%;)
35.茯苓酸a对照品(批号:10015,上海诗丹德,纯度:98.1%;)
36.试剂:乙腈为色谱纯,磷酸、甲醇为分析纯,水为纯化水。
37.2.色谱条件与系统适用性试验
38.以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,0.3%磷酸溶液为流动相b,按表1中的规定进行梯度洗脱;柱温为30℃;流速为每1.0ml/min,检测波长为243nm。理论板数按茯苓酸b峰计算应不低于10000。
39.表1洗脱条件
40.时间(min)流动相a(%)流动相b(%)0~3555
→
6345
→
3735-3663
→
5537
→
4536~455545
41.3.实验方法与结果
42.3.1测定方法
43.(1)供试品溶液的制备:取茯苓丁打粉后,过五号筛,精密称定2.0g,置锥形瓶中,精密加入体积分数为90%甲醇水溶液,称定重量,超声(250w,40khz)提取0.5h,放冷,用体积分数为90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得;
44.(2)对照品溶液的制备:为取茯苓酸a对照品、茯苓酸b对照品适量,精密称定,加适量甲醇溶解,制成每1ml含茯苓酸a 20μg和茯苓酸b 20μg的混合对照溶液;
45.(3)分别精密吸取对照品溶液和供试品溶液各20μl,注入液相色谱仪,按外标法以峰面积计算供试品中茯苓酸a和茯苓酸b的含量。
46.3.2提取溶剂的考察
47.取1.0g茯苓丁样品粉末(过五号筛),精密称定4份,置棕色具塞锥形瓶中,分别加入50%甲醇、70%甲醇、90%甲醇、纯甲醇20ml,称定重量,振摇2h、超声提取1h,放冷,分别用50%甲醇、70%甲醇、90%甲醇、甲醇补足减失的重量,摇匀,滤过,即得供试品溶液,注入高效液相色谱仪测定,分别计算茯苓酸b、茯苓酸a的含量,表2结果表明,提取溶剂为90%甲醇时,茯苓中茯苓酸b、茯苓酸a的含量较高,故选择提取溶剂为90%甲醇。
48.表2提取溶剂的考察
49.提取溶剂茯苓酸b的含量(%)茯苓酸a的含量(%)50%甲醇0.00320.002170%甲醇0.00970.009090%甲醇0.01010.0096纯甲醇0.00980.0092
50.3.3提取方式考察
51.取1.0g茯苓丁样品粉末(过五号筛),精密称定2份,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,分别振摇2h,超声1h、振摇2h,加热回流1h,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。注入液相色谱仪进行测定,分别计
算茯苓酸b、茯苓酸a的含量,表3结果表明,在不对比提取时间的情况下,提取方式为振摇2h,超声1h时,茯苓中茯苓酸b、茯苓酸a的含量较高。
52.表3提取方式考察
53.提取方式茯苓酸b的含量(%)茯苓酸a的含量(%)振摇2h,超声1h0.00920.0090振摇2h,加热回流1h0.00910.0088
54.3.4提取时间考察
55.取1.0g茯苓丁样品粉末(过五号筛),精密称定7份,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,分别按表4中不同时间超声提取,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。注入液相色谱仪进行测定,分别采用外标法计算茯苓酸b、茯苓酸a的含量,表4结果表明,不同提取时间对茯苓酸b和茯苓酸a含量影响不大,考虑效率和成本的情况下,最终选择提取方式和时间为超声提取0.5h。
56.表4提取时间考察
57.提取时间茯苓酸b的含量(%)茯苓酸a的含量(%)振摇2h,超声1h0.01080.0093振摇1.5h,超声1h0.01080.0091振摇1h,超声1h0.01090.0092振摇1.5h,超声0.5h0.01070.0092振摇0.5h,超声0.5h0.01080.0092超声1h0.01090.0094超声0.5h0.01080.0093
58.3.5称样量考察
59.分别取茯苓丁样品粉末(过五号筛)0.5g、1.0g、1.5g、2.0g,精密称定,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声提取30min,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。注入液相色谱仪进行测定,分别计算茯苓酸b、茯苓酸a的含量,表5结果表明,称样量为2.0g时,茯苓中茯苓酸b、茯苓酸a的含量较高,故选择称样量为2.0g。
60.表5称样量考察
61.称样量(g)茯苓酸b的含量(%)茯苓酸a的含量(%)0.50.01110.00941.00.01100.00941.50.01110.00952.00.01130.0095
62.4.方法学验证
63.4.1专属性
64.以90%甲醇溶液为阴性对照溶液;取上述供试品溶液、茯苓酸b对照品溶液、茯苓酸a对照品溶液、阴性对照溶液按上述检测方法获得其色谱图为图1,图1结果表明,阴性对照品溶液在茯苓酸b、茯苓酸a保留时间处无色谱峰,且茯苓供试品溶液中茯苓酸b、茯苓酸a
与相邻杂质峰分离度较好,方法专属性良好。
65.4.2标准曲线的绘制
66.4.2.1茯苓酸b标准曲线
67.茯苓酸b对照品贮备液
①
制备:精密称取茯苓酸b对照品6.49mg,至20ml棕色容量瓶中,加甲醇稀释至刻度,摇匀,即得。
68.茯苓酸b对照品贮备液
②
制备:精密量取茯苓酸b对照品贮备液
①
3.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,即得。
69.茯苓酸b线性溶液
①
制备:精密量取茯苓酸b对照品贮备液
②
0.5ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
70.茯苓酸b线性溶液
②
制备:精密量取茯苓酸b对照品贮备液
②
1.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
71.茯苓酸b线性溶液
③
制备:精密量取茯苓酸b对照品贮备液
②
2.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
72.茯苓酸b线性溶液
④
制备:精密量取茯苓酸b对照品贮备液
②
5.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
73.茯苓酸b线性溶液
⑤
制备:取茯苓酸b对照品贮备液
②
滤过,即得;
74.精密量取茯苓酸b线性溶液
①
~
⑤
各20μl,注入高效液相色谱仪,测定茯苓酸b色谱峰峰面积,以茯苓酸b色谱峰的峰面积为纵坐标,茯苓酸b的浓度为横坐标,进行线性回归,详见表6、图2,求得回归方程为y=30.749x+10.983,r2=0.9999,其线性范围为4.77μg/ml~95.40μg/ml,线性关系良好,可以采用外标一点法定量。
75.表6茯苓酸b标准曲线数据
76.溶液名称茯苓酸b浓度(μg/ml)峰面积线性溶液
①
4.77150.64线性溶液
②
9.54298.21线性溶液
③
19.08608.37线性溶液
④
47.701484.89线性溶液
⑤
95.402939.61
77.4.2.2茯苓酸a标准曲线
78.茯苓酸a对照品贮备液
①
制备:精密称取茯苓酸a对照品8.27mg,至25ml棕色容量瓶中,加甲醇稀释至刻度,摇匀,即得。
79.茯苓酸a对照品贮备液
②
制备:精密量取茯苓酸a对照品贮备液
①
3.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,即得。
80.茯苓酸a线性溶液
①
制备:精密量取茯苓酸a对照品贮备液
②
0.5ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
81.茯苓酸a线性溶液
②
制备:精密量取茯苓酸a对照品贮备液
②
1.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
82.茯苓酸a线性溶液
③
制备:精密量取茯苓酸a对照品贮备液
②
2.0ml置10ml棕色容量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
83.茯苓酸a线性溶液
④
制备:精密量取茯苓酸a对照品贮备液
②
5.0ml置10ml棕色容
量瓶中,用甲醇稀释至刻度,摇匀,滤过,即得;
84.茯苓酸a线性溶液
⑤
制备:取茯苓酸a对照品贮备液
②
滤过,即得;
85.精密量取茯苓酸a线性溶液
①
~
⑤
各20μl,注入高效液相色谱仪,测定茯苓酸a色谱峰峰面积,以茯苓酸a色谱峰的峰面积为纵坐标,茯苓酸a的浓度为横坐标,进行线性回归,详见表7、图3,求得回归方程为y=27.003x-7.4206,r2=1.0000,其线性范围为4.87μg/ml~97.354μg/ml,线性关系良好,可以采用外标一点法定量。
86.表7茯苓酸a标准曲线数据
87.溶液名称茯苓酸a浓度(μg/ml)峰面积线性溶液
①
4.87124.72线性溶液
②
9.74253.17线性溶液
③
19.47519.01线性溶液
④
48.681309.13线性溶液
⑤
97.3542620.47
88.4.3重复性试验
89.取2.0g茯苓丁样品粉末(过五号筛),精密称定,取6份,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声提取30min,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液,注入液相色谱仪进行测定,结果见表8,结果表明,茯苓酸b含量的rsd为0.5%,茯苓酸a含量的rsd为1.2%,重复性良好,符合分析要求。
90.表8茯苓酸b、茯苓酸a重复性试验结果
91.序号茯苓酸b含量(%)茯苓酸a含量(%)10.01100.008820.01110.008630.01120.008540.01110.008650.01110.008560.01120.0086平均值0.01110.0086rsd(%)0.51.2
92.4.4中间精密度
93.不同分析人员在不同时间利用另一台waters高效液相色谱仪,进行重复性实验。取2.0g本品粉末(过五号筛),精密称定,取6份,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声提取30min,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液,注入液相色谱仪进行测定。结果见表9,结果表明,茯苓酸b含量的rsd为0.7%,不同仪器测定茯苓中茯苓酸b含量的rsd为1.4%,茯苓酸a含量的rsd为1.6%,不同仪器测定茯苓中茯苓酸a含量的rsd为2.3%,符合分析要求。
94.表9中间精密度数据
95.序号茯苓酸b含量(%)茯苓酸a含量(%)10.01080.0088
20.01080.008830.01090.008840.01100.009250.01080.008960.01080.0088平均值0.01080.0089rsd(%)0.71.6不同仪器rsd(%)1.42.3
96.4.5回收率试验
97.回收率贮备液制备:精密量取茯苓酸b对照品贮备液
①
3.4ml、茯苓酸a对照品贮备液
①
2.7ml于200ml棕色量瓶,用90%甲醇稀释至刻度,摇匀即得。
98.取茯苓丁样品粉末(过五号筛)约2.0g,精密称定6份,置棕色具塞锥形瓶中,精密加入回收率贮备液20ml,称定重量,超声提取30min,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得加样回收率溶液。注入液相色谱仪进行测定。结果见表10、表11,结果表明,茯苓酸b的加样回收率在86.98%~91.40%之间,其回收率均值为89.13%,rsd为1.9%,茯苓酸a的加样回收率范围在85.14%~96.59%之间,其回收率均值为90.58%,rsd为4.7%,回收率良好,符合分析要求。计算公式如下式。
[0099][0100]
表10茯苓酸b回收率试验
[0101][0102]
表11茯苓酸a回收率试验
[0103][0104]
4.6稳定性考察
[0105]
取2.0g茯苓丁样品粉末(过五号筛),精密称定,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声提取30min,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液,注入液相色谱仪进行测定。分别在第0h、2h、4h、8h、12h、24h进
样测定同一供试品溶液中茯苓酸b和茯苓酸a的含量,计算rsd值,结果见表12,结果表明,茯苓酸b含量的rsd为1.3%,茯苓酸a含量的rsd为2.4%,供试品溶液稳定性良好,符合分析要求。
[0106]
表12样品稳定性试验
[0107]
测定时间(h)茯苓酸b含量(%)茯苓酸a含量(%)00.01050.008220.01030.008440.01060.008680.01040.0087120.01060.0084240.01060.0087平均值0.01050.0085rsd(%)1.32.4
[0108]
4.7耐用性考察
[0109]
取2.0g茯苓丁样品粉末(过五号筛),精密称定,置棕色具塞锥形瓶中,精密加入90%甲醇20ml,称定重量,超声提取30min,放冷,用90%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液,注入液相色谱仪进行测定。
[0110]
采用不同色谱柱、不同流速(1.0
±
0.1ml/min)、不同柱温(30
±
5℃)测定同一供试品溶液,考察实验方法对不同色谱柱、不同流速、不同柱温的耐用性。结果见表13,结果表明,测得茯苓酸b的含量rsd值为1.0%,茯苓酸a的含量rsd值为2.8%,符合分析要求。表明该方法对不同色谱柱、不同流速、不同柱温耐用性较好。
[0111]
表13不同色谱柱、不同流速、不同柱温含量测定
[0112][0113]
上述实验表明,该方法可为检测茯苓配方颗粒所用茯苓药材和饮片提供参考,本方法两个成分理论塔板数均大于10000,成分保留及洗脱能力佳,方便快捷成本低,便于普及应用。
[0114]
以上所述,仅为本技术的实施例而已,本技术的保护范围并不受这些具体实施例的限制,而是由本技术的权利要求书来确定。对于本领域技术人员来说,本技术可以有各种
更改和变化。凡在本技术的技术思想和原理之内所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1