一种小儿复方氨基酸注射液中有关物质的检测方法与流程
1.本发明涉及药物检测技术领域,具体涉及一种小儿复方氨基酸注射液中有关物质的检测方法。
背景技术:
2.小儿复方氨基酸注射液(19aa-i)主要适用于小儿患消化系统疾病,不能经肠胃摄取食物者,由各种疾病所引起的低蛋白血症者,受严重创伤、烧伤及败血症等体内氮平衡失调者。小儿复方氨基酸注射液的有效成分包含异亮氨酸、亮氨酸、醋酸赖氨酸、甲硫氨酸、苯丙氨酸、苏氨酸、缬氨酸、盐酸半胱氨酸、组氨酸、酪氨酸、乙酰酪氨酸、丙氨酸、精氨酸、脯氨酸、丝氨酸、门冬氨酸、谷氨酸、甘氨酸、牛磺酸和色氨酸二十种氨基酸。然而,在光照条件下,小儿复方氨基酸注射液中的色氨酸会发生降解,产生色氨酸降解杂质(包括色氨酸杂质b、色氨酸杂质c、色氨酸杂质d和色氨酸杂质e),影响小儿复方氨基酸注射液的安全使用。因此,控制小儿复方氨基酸注射液中色氨酸降解杂质(有关物质)的含量至关重要。
3.然而,小儿复方氨基酸注射液成分复杂,各组分极性差别较大,干扰因素多,无法同时测定小儿复方氨基酸注射液中色氨酸降解杂质。
技术实现要素:
4.鉴于此,本发明的目的在于提供一种小儿复方氨基酸注射液中有关物质的检测方法。本发明所述的检测方法能够检测出小儿复方氨基酸注射液中有关物质(色氨酸降解杂质),且该检测方法的灵敏度、准确度高。
5.为了实现上述发明目的,本发明提供一种小儿复方氨基酸注射液中有关物质的检测方法,包括以下步骤:
6.采用高效液相色谱对待测样品进行检测,得到待测样品的色谱图;
7.将所述待测样品的色谱图与预定的标准曲线进行对比,采用外标法计算得到待测样品中有关物质的含量;
8.所述高效液相色谱的检测条件包括:流动相体系包括流动相a和流动相b;所述流动相a为磷酸盐缓冲液-甲醇体系;所述流动相b为磷酸盐缓冲液-甲醇-乙腈体系;所述高效液相色谱的色谱柱为c
18
柱;
9.所述流动相体系的流速为1ml/min;
10.洗脱方式为梯度洗脱;
11.所述梯度洗脱的程序为:
12.0~22min:所述流动相a的体积百分含量为100%;
13.22~46min:所述流动相a的体积百分含量由100%匀速减少到0%;
14.46~60min:所述流动相a的体积百分含量为0%;
15.60~61min:所述流动相a的体积百分含量由0%匀速增加到100%;
16.61~70min:所述流动相a的体积百分含量为100%;
17.检测器为紫外检测器和pda检测器,检测波长为220nm;
18.所述有关物质为色氨酸降解杂质。
19.优选地,所述色氨酸降解杂质包括色氨酸杂质b、色氨酸杂质c、色氨酸杂质d和色氨酸杂质e。
20.优选地,所述流动相a中磷酸盐缓冲液为二水合磷酸二氢钠溶液。
21.优选地,所述二水合磷酸二氢钠溶液的浓度为3.5~4.3g/l。
22.优选地,所述流动相a中磷酸盐缓冲液和甲醇的体积比为94:6。
23.优选地,所述流动相b中磷酸盐缓冲液为二水合磷酸二氢钠溶液;所述二水合磷酸二氢钠溶液的浓度为3.5~4.3g/l。
24.优选地,所述流动相b中磷酸盐缓冲液、甲醇和乙腈的体积比为60:6:34。
25.优选地,所述高效液相色谱的柱温为36~45℃;所述高效液相色谱的进样量为20μl。
26.优选地,所述流动相体系的ph值为5.0。
27.本发明提供了一种小儿复方氨基酸注射液中有关物质的检测方法,包括以下步骤:
28.本发明采用高效液相色谱对待测样品进行检测,得到待测样品的色谱图;将所述待测样品的色谱图与预定的标准曲线进行对比,采用外标法计算得到待测样品中有关物质的含量;所述高效液相色谱的检测条件包括:流动相体系包括流动相a和流动相b;所述流动相a为磷酸盐缓冲液-甲醇体系;所述流动相b为磷酸盐缓冲液-甲醇-乙腈体系;所述高效液相色谱的色谱柱为c
18
柱;所述流动相体系的流速为1ml/min;洗脱方式为梯度洗脱;所述梯度洗脱的程序为:0~22min:所述流动相a的体积百分含量为100%;22~46min:所述流动相a的体积百分含量由100%匀速减少到0%;46~60min:所述流动相a的体积百分含量为0%;60~61min:所述流动相a的体积百分含量由0%匀速增加到100%;61~70min:所述流动相a的体积百分含量为100%;检测器为紫外检测器和pda检测器,检测波长为220nm;所述有关物质为色氨酸降解杂质。本发明通过限定高效液相色谱的检测的色谱柱以及洗脱方式和具体洗脱程序,采用非极性固定相和极性流动相梯度变化至中等极性的液相色谱体系,利用反相色谱分离极性相近的色氨酸降解杂质,避免了小儿复方氨基酸注射液(19aa-i)中的其他成分的干扰,能够准确检测出儿复方氨基酸注射液中有关物质。且该检测方法的灵敏度、准确度和精确度高。
附图说明
29.图1为色氨酸杂质b的线性图谱;
30.图2为色氨酸杂质c的线性图谱;
31.图3为色氨酸杂质d的线性图谱;
32.图4为色氨酸杂质e的线性图谱;
33.图5为色氨酸的线性图谱;
34.图6为空白溶液的色谱图;
35.图7为空白辅料溶液的色谱图;
36.图8为加标供试品溶液的色谱图。
具体实施方式
37.本发明提供了一种小儿复方氨基酸注射液中有关物质的检测方法,包括以下步骤:
38.采用高效液相色谱对所述待测样品进行检测,得到待测样品的色谱图;
39.将所述待测样品的色谱图与预定的标准曲线进行对比,采用外标法计算得到待测样品中有关物质的含量。
40.在本发明中,所述高效液相色谱的检测条件包括:流动相体系包括流动相a和流动相b;所述流动相a为磷酸盐缓冲液-甲醇体系;所述流动相b为磷酸盐缓冲液-甲醇-乙腈体系;所述高效液相色谱的色谱柱为c
18
柱;
41.所述流动相体系的流速为1ml/min;
42.洗脱方式为梯度洗脱;
43.所述梯度洗脱的程序为:
44.0~22min:所述流动相a的体积百分含量为100%;
45.22~46min:所述流动相a的体积百分含量由100%匀速减少到0%;
46.46~60min:所述流动相a的体积百分含量为0%;
47.60~61min:所述流动相a的体积百分含量由0%匀速增加到100%;
48.61~70min:所述流动相a的体积百分含量为100%;
49.检测器为紫外检测器和pda检测器,检测波长为220nm。
50.在本发明中,若没有特殊说明,所采用的试剂均为本领域技术人员所熟知的市售商品。
51.在本发明中,所述有关物质为色氨酸降解杂质,所述色氨酸降解杂质优选包括色氨酸杂质b、色氨酸杂质c、色氨酸杂质d和色氨酸杂质e,优选地,所述色氨酸降解杂质还包括少量未知色氨酸降解杂质。
52.本发明中,所述流动相体系的ph值优选为5.0。在本发明中,所述流动相a中的磷酸盐缓冲液优选为二水合磷酸二氢钠溶液,所述二水合磷酸二氢钠溶液的浓度优选为3.5~4.3g/l,更优选为3.90g/l。在本发明中,所述流动相a中磷酸盐缓冲液和甲醇的体积比优选为94:6。
53.在本发明中,所述流动相b中的磷酸盐缓冲液优选为二水合磷酸二氢钠溶液,所述二水合磷酸二氢钠溶液的浓度优选为3.5~4.3g/l,更优选为3.90g/l。在本发明中,所述流动相b中磷酸盐缓冲液、甲醇和乙腈的体积比优选为60:6:34。
54.本发明采用非极性固定相和极性流动相梯度变化至中等极性的液相色谱体系,利用反相色谱分离极性相近的色氨酸降解杂质,避免了小儿复方氨基酸注射液(19aa-i)中的其他成分的干扰。
55.在本发明中,所述c
18
柱优选为watersatlantis t3柱(4.6mm
×
250mm,5μm)。在本发明中,所述高效液相色谱的色谱柱的柱温优选为36~45℃,更优选为40℃。在本发明中,所述高效液相色谱的进样量优选为20μl。
56.在本发明中,所述检测器为紫外检测器和pda检测器,检测波长为220nm。在本发明中,所述紫外检测器优选为2489紫外检测器。在本发明中,所述pda检测器优选为2998pda检测器。
57.在本发明中,所述预定的标准曲线的的获取方法优选包括:
58.分别将色氨酸杂质b对照品贮备溶液、色氨酸杂质c对照品贮备溶液、色氨酸杂质d对照品贮备溶液、色氨酸杂质e对照品贮备溶液系列标准溶液和色氨酸对照品贮备溶液系列标准溶液,按照高效液相色谱的检测方法进行测定,得到色氨酸杂质b~e对照品和色氨酸对照品的色谱图;
59.分别以色氨酸杂质b~e对照品和色氨酸对照品色谱图中的峰面积为横坐标,浓度为纵坐标,分别得到色氨酸杂质b~e对照品和色氨酸对照品的预定的标准曲线。
60.在本发明中,上述检测条件下,色氨酸杂质b对照品、色氨酸杂质c对照品、色氨酸杂质d对照品、色氨酸杂质e对照品和色氨酸对照品的出峰时间分别优选为6.756min、7.835min、10.533min、9.17min、14.497min、22.099min。在本发明中,所述色氨酸杂质b~e对照品优选购自trc公司;所述色氨酸对照品优选购自中国食品药品检定研究院公司。
61.在本发明中,所述色氨酸对照品用于定量少量未知色氨酸降解杂质。
62.在本发明中,所述外标法优选包括将所述待测样品进行检测,所得待测样品的色谱图中的峰面积代入对应杂质的预定的标准曲线中,计算得到待测样品中有关物质的含量。
63.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
64.在本发明的实施例中,高效液相色谱的检测条件如下所示:
65.高效液相色谱仪为waters 2695;色谱柱为:waters atlantis t3(4.6mm
×
250mm,5μm);柱温为40℃;进样量为20μl;流动相a为磷酸盐缓冲液(3.90g/l的二水合磷酸二氢钠溶液)-甲醇体系(体积比为94:6);流动相b为磷酸盐缓冲液(3.90g/l的二水合磷酸二氢钠溶液)-甲醇-乙腈体系(体积比为60:6:34);流动相体系的流速为1ml/min;洗脱方式为梯度洗脱;梯度洗脱程度为:0~22min:流动相a的体积百分含量为100%;22~46min:流动相a的体积百分含量由100%匀速减少到0%;46~60min:流动相a的体积百分含量为0%;60~61min:流动相a的体积百分含量由0%匀速增加到100%;61~70min:流动相a的体积百分含量为100%;检测器为2489紫外检测器和2998pda检测器;检测波长为220nm。所述色氨酸杂质b~e对照品购自trc公司,色氨酸对照品购自中国食品药品检定研究院公司。
66.分析天平:mettlerxp 205。
67.实施例1
68.一、工作液的配置
69.1、有关物质对照品贮备液的配制
70.色氨酸杂质b对照品贮备液:精密称取色氨酸杂质b对照品4.5mg,置25ml量瓶中,加水超声至溶解,用水稀释至刻度,摇匀,得到色氨酸杂质b对照品贮备液。
71.色氨酸杂质c对照品贮备液:精密称取色氨酸杂质c对照品4.5mg,置25ml量瓶中,加水超声至溶解,用水稀释至刻度,摇匀,得到色氨酸杂质c对照品贮备液。
72.色氨酸杂质d对照品贮备液:精密称取色氨酸杂质d对照品4.5mg,置25ml量瓶中,加水超声至溶解,用水稀释至刻度,摇匀,得到色氨酸杂质d对照品贮备液。
73.色氨酸杂质e对照品贮备液:精密称取色氨酸杂质e对照品4.5mg,置25ml量瓶中,加水超声至溶解,用水稀释至刻度,摇匀,得到色氨酸杂质e对照品贮备液。
74.色氨酸对照品贮备液:精密称取色氨酸对照品3.0mg,置25ml量瓶中,加水超声至溶解,用水稀释至刻度,摇匀,作为色氨酸对照品贮备液。
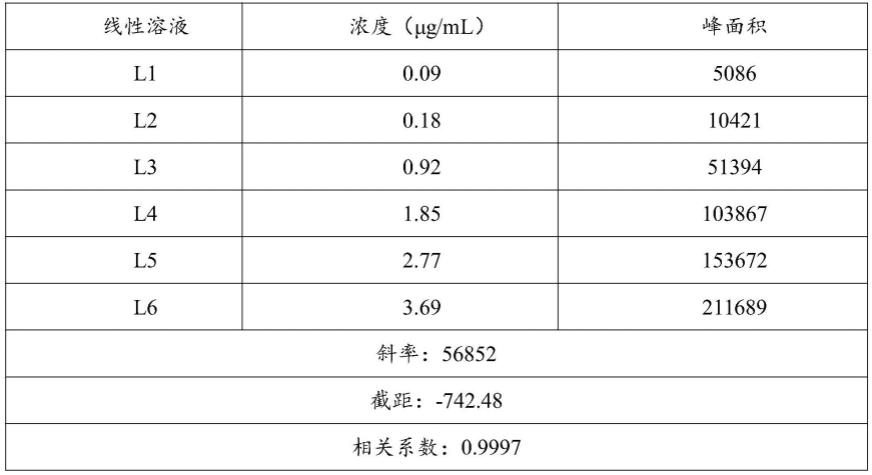
75.2、混合对照品溶液的配制
76.精密量取色氨酸杂质b对照品、色氨酸杂质c对照品、色氨酸杂质d对照品、色氨酸杂质e对照品、色氨酸对照品贮备液各1ml,置于同一100ml量瓶中,加水稀释至刻度,摇匀,得到混合对照品溶液。
77.3、灵敏度溶液的配制
78.精密量取混合对照品溶液1ml,置于25ml量瓶中,用水稀释至刻度,摇匀,得到灵敏度溶液。
79.4、系统适用性溶液
80.精密量取色氨酸杂质c对照品贮备液0.1ml,置于10ml量瓶中,用本品稀释至刻度,摇匀,得到系统适用性溶液。
81.5、空白辅料溶液的配制:精密称取异亮氨酸491.3mg、亮氨酸840.1mg、醋酸赖氨酸489.7mg、甲硫氨酸201.7mg、苯丙氨酸290.5mg、苏氨酸250.8mg、缬氨酸473.1mg、盐酸半胱氨酸20.02mg、组氨酸297.1mg、酪氨酸41.04mg、乙酰酪氨酸121.7mg、丙氨酸321.1mg、精氨酸728.9mg、脯氨酸411.3mg、丝氨酸233.0mg、门冬氨酸191.3mg、谷氨酸308.6mg、甘氨酸221.9mg、牛磺酸15.17mg、亚硫酸氢钠50.55mg,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,作为空白辅料溶液。
82.6、色氨酸杂质b定位溶液:将色氨酸杂质b对照品贮备液用水稀释,至浓度为45μg/ml,得到色氨酸杂质b定位溶液。
83.8、色氨酸杂质c定位溶液:将色氨酸杂质c对照品贮备液用水稀释,至浓度为45μg/ml,得到色氨酸杂质c定位溶液。
84.9、色氨酸杂质d定位溶液:将色氨酸杂质d对照品贮备液用水稀释,至浓度为45μg/ml,得到色氨酸杂质d定位溶液。
85.10、色氨酸杂质e定位溶液:将色氨酸杂质e对照品贮备液用水稀释,至浓度为45μg/ml,得到色氨酸杂质e定位溶液。
86.11、空白溶液的配制:去离子水25ml。
87.12、供试品溶液的配制:小儿复方氨基酸注射液(19aa
‑ⅰ
)25ml。
88.13、加标供试品溶液的配制:精密量取色氨酸杂质b对照品、色氨酸杂质c对照品、色氨酸杂质d对照品、色氨酸杂质e对照品、色氨酸对照品贮备液各1ml,空白辅料贮备液5ml,置于同一100ml量瓶中,用水稀释至刻度,摇匀。
89.二、检测
90.1、线性标准曲线
91.分别精密量取色氨酸杂质b~e对照品贮备溶液,用水进行稀释制成含各杂质浓度为0.09、0.18、0.90、1.80、2.70、3.60μg/ml的系列标准溶液和含色氨酸0.06、0.12、0.60、1.20、1.80、2.40μg/ml的系列标准溶液,取系列标准溶液20μl进样,按照高效液相色谱的检测方法进行测定,检测结果见图1~5和表1~5,从图1~5可知:色氨酸杂质b的线性方程为y
=56852x-742.48(r=0.9997),表明色氨酸杂质b在0.09~3.69μg/ml浓度范围内线性良好;色氨酸杂质c的线性方程为y=107299x+264.97(r=1.0000),表明色氨酸杂质c在0.09~3.59μg/ml范围内线性良好;色氨酸杂质d的线性方程为y=109380x-381.99(r=1.0000),表明色氨酸杂质d在0.08~3.23μg/ml范围内线性良好;色氨酸杂质e的线性方程为y=57802x-434.03(r=0.9999),表明色氨酸杂质e在0.09~3.59μg/ml范围内线性良好;色氨酸的线性方程为y=197148x+3282.4(r=0.9999),表明色氨酸在0.06~2.53μg/ml范围内线性良好。
92.表1色氨酸杂质b线性试验结果统计表
[0093][0094]
表2色氨酸杂质c线性试验结果统计表
[0095][0096]
表3色氨酸杂质d线性试验结果统计表
[0097][0098]
表4色氨酸杂质e线性试验结果统计表
[0099][0100][0101]
表5色氨酸线性试验结果统计表
[0102][0103]
2、专属性测试
[0104]
精密量取空白溶液、空白辅料溶液、色氨酸杂质b~e定位溶液、混合对照品溶液、加标供试品溶液各20μl,按照高效液相色谱的检测条件进行检测,得到色谱图,见图6~8,从图6~8可以得知:空白溶液及空白辅料溶液不干扰色氨酸杂质b、色氨酸杂质c、色氨酸杂质d及色氨酸杂质e的测定,表明该方法专属性较好。
[0105]
3、准确度测试
[0106]
分别精密量取色氨酸杂质b~e对照品贮备溶液和色氨酸对照品贮备溶液,加空白辅料贮备液,用水配制成杂质浓度分别为1.44μg/ml、1.80μg/ml和2.5μg/ml的色氨酸杂质b~e回收率溶液;色氨酸浓度分别为0.96μg/ml、1.20μg/ml和1.44μg/ml的色氨酸回收率溶液,分别取各回收率溶液20μl按照高效液相色谱的方法进行测定,测定结果为:色氨酸杂质b的回收率为91.31~97.52%,rsd%为2.48%;色氨酸杂质c的回收率为95.62~96.83%,rsd%为0.39%;色氨酸杂质d的回收率为97.83~98.94%,rsd%为0.41%;色氨酸杂质e的回收率为101.3~104.1%,rsd%为1.06%;色氨酸的回收率为98.56~100.4%,rds为0.67%。表明该方法准确度较好。
[0107]
4、重复性和中间精密度测试
[0108]
分别精密量取色氨酸杂质b~e对照品贮备溶液,加空白辅料贮备液,用水配制成杂质浓度为1.80μg/ml,色氨酸浓度为1.20μg/ml的100%回收率溶液6份,分别取各回收率溶液20μl进样,按高效液相色谱的检测条件进行测定。重复性结果为:6份中间精密度样品溶液中色氨酸杂质b、色氨酸杂质c、色氨酸杂质d、色氨酸杂质e和色氨酸的平均回收率分别为94.22%、98.73%、97.62%、101.0%和96.76%,rsd%分别为1.98%、0.16%、0.17%、0.30%和0.51%;中间精密度结果为:6份样品的色氨酸杂质b~e和色氨酸的平均回收率分别为94.22%、98.73%、97.62%、101.0%和96.76%,rsd%分别为1.98%、0.16%、0.17%、0.30%和0.51%。表明该方法精密度较好。
[0109]
5、定量限与检测限
[0110]
分别精密量取色氨酸杂质b~e对照品贮备溶液,用水进行稀释制成信噪比约为10的定量限溶液及信噪比约3的检测限溶液,分别取各定量限溶液及检测限溶液20μl按照高
效液相色谱的检测条件进行检测。结果显示色氨酸色氨酸杂质b~e和色氨酸的定量限分别为22.16ng/ml、7.90ng/ml、8.39ng/ml、21.51ng/ml和7.58ng/ml,检测限分别为13.85ng/ml、4.31ng/ml、4.52ng/ml、10.76ng/ml和3.54ng/ml;表明该方法的检测灵敏度较高。
[0111]
实施例2
[0112]
取小儿复方氨基酸注射液(19aa-i)(华润双鹤药业股份有限公司生产)的3批样品进行检测,色氨酸杂质b~e均小于0.05%,未知杂质以色氨酸计均小于0.10%。
[0113]
注:供试品溶液色谱图中如有杂质峰,杂质b~e按外标法,以峰面积计算,代入杂质b~e标准曲线,得到含量,杂质b~e的含量不得超过色氨酸标示量的0.15%;其他色氨酸杂质按外标法,以峰面积计算,代入色氨酸标准曲线,得到其他色氨酸杂质的含量,不得过色氨酸标示量的0.10%,杂质总量(杂质b~e和其他色氨酸杂质)不得过色氨酸标示量的2.0%。
[0114]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1