一种小儿银连颗粒剂的质量检测方法与流程
1.本发明属于中药质量检测技术领域,涉及一种小儿银连颗粒剂的质量检测方法,具体涉及所述小儿银连颗粒剂的药材鉴别、特征图谱检测及其有效成分含量测定方法。
背景技术:
2.流行性感冒是由流行性感冒病毒引起的急性呼吸道疾病,具有强烈传染性和快速传播性,其高危人群多为儿童。流行病学研究结果显示,近年来儿童流行性感冒患病率>60%。儿童免疫功能低下,尤其是婴幼儿,极易受到流行性感冒病毒侵袭,相较于成人更易患呼吸道传染病。
3.目前,由金银花、黄连和灯心草制成的中药复方制剂是一种清热解毒药,用于降心火祛胎毒、祛血瘀、心烦、失眠、小儿夜啼。该中药复方制剂以黄连为主药,它具有清热泻火,燥湿、解毒,辅以金银花、灯心草起协同作用,以增加黄连效用,对风热感冒(急性上呼吸道感染)、流行性感冒均有很好的疗效。
4.专利文献cn104784372a公开了一种治疗心热内扰型夜啼的中药复方制剂,该治疗心热内扰型夜啼的中药复方制剂由金银花2~40份,黄连2~40份,灯心草2~40份组成。各组分可以分别提取后再混合,也可以混合后再提取;提取溶剂可以是水或10~95%的乙醇;提取方法是常规的中药提取方法。所制备的药物组合物可以用常规方法制成各种口服制剂,制得的中药复方制剂具有清热解毒的作用,对降心火祛胎毒、祛血淤,心烦、失眠、小儿夜啼等症状具有很好的治疗效果。
5.专利文献cn106860695a公开了一种治疗心热内扰型夜啼的中药复方制剂,该治疗心热内扰型夜啼的中药复方制剂由金银花5~60份,黄连5~60份,灯心草5~60份组成。该治疗夜啼的中药复方制剂,因为处方的科学合理的配伍,不同的提取方法均可达到理想的治疗效果。
6.现有的由金银花、黄连和灯心草制成的口服制剂目前还未见有相关的质量检测报道,而药品安全,尤其是药物成分复杂的中药复方制剂的安全性问题一直是消费者首要关注的问题。因此,研究和发开出一种由金银花、黄连和灯心草制成的中药复方制剂的质量检测标准是目前亟需解决的问题。
技术实现要素:
7.为了解决现有技术中存在的缺陷,本发明提供了一种小儿银连颗粒剂的质量检测方法。该小儿银连颗粒剂是由金银花、黄连和灯心草3味药材组成的复方,为了保证该颗粒剂在临床上的疗效与质量稳定,本发明提供的质量检测方法除了对复方中的绿原酸和小檗碱进行了含量测定,对灯心草进行了鉴别外,还建立了特征图谱,全面地反映该颗粒剂中所含化学成分的种类与数量,弥补了单纯指标性成分在质控方面的不足和薄层鉴别的模糊性,更具有科学性与全面性,保证了中药质量的均一性和稳定性,提高了在临床上的整体疗效。
8.本发明提供了一种小儿银连颗粒剂的质量检测方法,所述小儿银连颗粒剂由黄连400g、金银花400g、灯心草400g、β-环糊精480g、三氯蔗糖60g、硬脂酸镁5g和香精10g,蔗糖粉加至1000g制成,其质量检测方法包括药材鉴别、特征图谱检测和含量测定方法:
9.一、灯心草的薄层鉴定:
10.1.1、取样品2.5g,研细,加甲醇20ml,超声处理20min,滤过,滤液蒸干,残渣加水20ml溶解,过固相萃取小柱,弃去流出液,加甲醇1ml洗脱定容,作为供试品溶液;
11.1.2、另取灯心草药材1g,加甲醇100ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣用乙醚2ml洗涤,弃去乙醚液,加甲醇1ml使溶解,作为对照药材溶液;
12.1.3、吸取上述供试品溶液4~8μl、对照药材溶液2μl,分别点于同一硅胶g薄层板上,加入展开剂,预饱和后展开,取出,晾干,置紫外光灯下检视,供试品色谱中在254nm波长下可检视出与对照药材相应斑点;
13.二、特征图谱检测:
14.2.1、色谱条件与系统适用性试验
15.以十八烷基硅烷键合硅胶为填充剂(waters acquity uplc beh c18色谱柱,50
×
2.1mm,1.7μm);以乙腈为流动相a,以含0.05%十二烷基磺酸钠的0.1%磷酸水溶液为流动相b,流速:0.4ml/min;检测波长为327nm,理论板数按绿原酸峰计算应不低于3000;
16.2.2、对照品溶液的制备
17.取盐酸小檗碱、绿原酸对照品,精密称定,加甲醇制成每1ml各含40μg的溶液,得对照品溶液;
18.2.3、供试品溶液的制备
19.取样品,研细,取0.5g,精密称定,置具塞锥形瓶中,精密加入50%甲醇50ml,称定重量,超声处理30min,取出,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,得供试品溶液;
20.2.4、测定法
21.分别精密吸取参照品溶液与供试品溶液各1~2μl,注入超高效液相色谱仪,测定,记录色谱图,即得;
22.三、小檗碱和绿原酸的含量测定
23.3.1、色谱条件与系统适用性试验
24.以十八烷基硅烷键合硅胶为填充剂(waters acquity uplc beh c18色谱柱,50
×
2.1mm,1.7μm);以乙腈为流动相a,以含0.05%十二烷基磺酸钠的0.1%磷酸水溶液为流动相b,流速:0.4ml/min;检测波长为327nm,理论板数按绿原酸峰计算应不低于3000;
25.3.2、对照品溶液的制备
26.取盐酸小檗碱、绿原酸对照品适量,精密称定,加甲醇制成每1ml各含40μg的溶液,得对照品溶液;
27.3.3、供试品溶液的制备
28.取本品,研细,取0.3g,精密称定,置具塞锥形瓶中,精密加入50%甲醇50ml,称定重量,超声处理30min,取出,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,得供试品溶液;
29.3.4、测定法
30.分别精密吸取对照品溶液与供试品溶液各1~2μl,注入液相色谱仪,测定,即得。
31.进一步地,所述小儿银连颗粒剂的制备方法为:
32.步骤s1:取金银花干燥花蕾或带初开的花,黄连干燥的根茎,润透后切薄片,晾干,灯心草干燥的茎髓,得原药材;
33.步骤s2:取1/10量灯心草,得原料a,将原料a加入体积浓度为60%的乙醇浸湿,所述乙醇的重量是原料a总重量的20倍,装入渗漉罐;
34.步骤s3:取金银花和黄连粉碎成最粗粉,得原料b,将原料b加入体积浓度为60%的乙醇浸润,所述乙醇的重量是原料b总重量的2倍,装入渗漉罐;
35.步骤s4:取剩余灯心草,得原料c,将原料c加入体积浓度为60%的乙醇浸湿,所述乙醇的重量是原料c总重量的20倍,装入渗漉罐;
36.步骤s5:往渗漉罐中加入体积浓度为60%的乙醇,所述乙醇的加入量是原料a、原料b和原料c生药总重量的10倍,每千克药材以每分钟2~3ml进行渗漉,收集渗漉液;
37.步骤s6:将经步骤s5制得的渗漉液减压回收乙醇并在温度为50~60℃的条件下浓缩至相对密度为1.08~1.12,得浸膏,将浸膏与β-环糊精加热溶解混合均匀,喷雾干燥,得干膏,将干膏与三氯蔗糖,硬脂酸镁,香精,蔗糖粉加入混合机混合18~22min,干压制粒,过筛,包装,即得。
38.进一步地,所述灯心草的薄层鉴定步骤1.1中固相萃取小柱的规格为:c18,250mg/6ml。
39.进一步地,所述灯心草的薄层鉴定步骤1.3中的展开剂由环己烷和乙酸乙酯按体积比(9:7)组成。
40.进一步地,所述步骤2.3和步骤3.3中的超声处理条件为:功率250w,频率40khz。
41.进一步地,所述小檗碱和绿原酸的含量测定步骤3.3中的供试品溶液还含有保护剂。
42.进一步地,所述保护剂的添加量与小儿银连颗粒剂样品的质量比为0.1:100。
43.进一步地,所述保护剂由精氨酸、抗坏血酸钠和脂肪酸甲酯聚氧乙烯醚组成。
44.进一步地,所述保护剂由精氨酸、抗坏血酸钠和脂肪酸甲酯聚氧乙烯醚按1:1:2组成。
45.本发明所述的药材:金银花为忍冬科植物忍冬(lonicera japonica thunb.)的干燥花蕾或带初开的花经加工所得的饮片;黄连为毛茛科植物黄连(coptis chinensis franch.)的干燥根茎;灯心草为灯心草科植物灯心草(juncus effusus l.)的干燥茎髓,经加工制得。所述药材前处理为:将金银花铺于净选工作台上,拣去非药用部位等杂质,筛选去泥沙、灰尘;将黄连铺于净选工作台上,拣去虫蛀、霉变、非药用部位,筛选除去泥沙等杂质,润透后切薄片,晾干,或用时捣碎;将灯心草铺于拣选工作台,除去杂质。本发明中没有限定体积浓度的甲醇为纯甲醇(100%),50%甲醇为体积浓度为50%的甲醇。
46.本发明所述喷雾干燥的条件为:进口温度190~210℃,出口温度70~90℃;所述过筛为过筛网16目,未成形颗粒的干粉经65目筛网筛分后,重复干压制粒,至细粉比例小于投料量5%;所述包装为:领取合格颗粒,确认环境湿度已处于在60%以下,分装颗粒,按每包3g调节装量调节器,使装量差异在标准范围内(2.79g~3.21g或3g
±
7%)。
47.本发明提供的小儿银连颗粒是由黄连、金银花、灯心草3味中药提取加工制成的中
药制剂,发明人参考《中国药典》2020年版有关药味鉴别项下的方法,对供试品制备方法、展开剂、显色条件、专属性、不同点样量、不同环境温度、不同环境湿度及不同厂家薄层板进行了考察,通过反复摸索,建立了以灯心草药材为对照的薄层鉴别。同时还建立了特征图谱,全面地反映该颗粒剂中所含化学成分的种类与数量,弥补了单纯指标性成分在质控方面的不足和薄层鉴别的模糊性。同时,由于灯心草无含量测定项指标性成分,黄连中3种代表性成分和金银花中5种代表性成分均已进行特征图谱的检测,选择小檗碱和绿原酸进行含量测定。
48.另外,由于绿原酸在环境中极其不稳定,申请人按以往的成功研究经验,在供试品溶液加入由精氨酸和维生素c组成的稳定剂用于提高检测结果的稳定性和重现性。但是,研究发现,其稳定效果也不佳,发明人猜测其原因是:可能是绿原酸氧化速度较快,该稳定剂没办法达到预期的稳定效果。发明人又添加了不同种类的抗氧剂进行联合使用,发现其效果仍然不佳。在一次摸索试验中,发明人意外地发现,添加一定量的脂肪酸甲酯聚氧乙烯醚可以提高精氨酸和维生素c对绿原酸的稳定性,可以大大提高检测结果的重现性。发明人猜测其原理可能是脂肪酸甲酯聚氧乙烯醚可以加快精氨酸和维生素c的扩散,促进精氨酸和维生素c的抗氧化作用,降低绿原酸的氧化反应,从而提高供试品溶液中绿原酸的稳定性。
49.总之,与现有技术相比,本发明提供的小儿银连颗粒剂的质量检测方法具有精密度高、稳定性好、重复性佳的优点,同时该质量检测方法具有科学性与全面性,保证了中药质量的均一性和稳定性,提高了在临床上的整体疗效。
附图说明:
50.图1为灯心草的薄层鉴定图;
51.图2为黄连的薄层鉴定图;
52.图3为金银花的薄层鉴定图;
53.图4为实施例1制得的颗粒剂供试品特征图谱图,其中:1为新绿原酸,2为绿原酸(s),3为隐绿原酸,4为阿魏酸,5为4,5-二-o-咖啡酰奎宁酸,6为黄连碱,7为巴马汀,8为盐酸小檗碱;
54.图5为缺金银花阴性样品色谱图;
55.图6为缺黄连阴性样品色谱图。
具体实施方式:
56.以下通过具体实施方式的描述对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以做出各种修改或改进,但是只要不脱离本发明的基本思想,均在本发明的范围之内。
57.实施例1、一种小儿银连颗粒剂
58.所述小儿银连颗粒剂由以下成分及其含量制成:
59.金银花400g、黄连400g、灯心草400g、β-环糊精450g、三氯蔗糖55g、硬脂酸镁4g和食用香精甜橙粉末8g,蔗糖粉加至1000g。
60.制备方法:
61.步骤s1:取金银花干燥花蕾或带初开的花,黄连干燥的根茎,润透后切薄片,晾干,
灯心草干燥的茎髓,得原药材;
62.步骤s2:取1/10量灯心草,得原料a,将原料a加入体积浓度为60%的乙醇浸湿,所述乙醇的重量是原料a总重量的20倍,装入渗漉罐;
63.步骤s3:取金银花和黄连粉碎成最粗粉,得原料b,将原料b加入体积浓度为60%的乙醇浸润,所述乙醇的重量是原料b总重量的2倍,装入渗漉罐;
64.步骤s4:取剩余灯心草,得原料c,将原料c加入体积浓度为60%的乙醇浸湿,所述乙醇的重量是原料c总重量的20倍,装入渗漉罐;
65.步骤s5:往渗漉罐中加入体积浓度为60%的乙醇,所述乙醇的加入量原料a、原料b和原料c生药总重量的10倍,每千克药材以每分钟2~3ml进行渗漉,收集渗漉液,
66.步骤s6:将经步骤s5制得的上清液减压回收乙醇并在温度为50℃的条件下浓缩至相对密度为1.08~1.12,得浸膏,将浸膏与β-环糊精加热溶解混合均匀,喷雾干燥,所述喷雾干燥的条件为:进口温度190℃,出口温度70℃,得干膏,将干膏与三氯蔗糖,硬脂酸镁,香精,蔗糖粉加入混合机混合18min,干压制粒,过筛,筛网16目,按每包3g包装,即得。
67.实施例2、小儿银连颗粒剂的药材薄层鉴定
68.灯心草对照药材、黄连对照药材购自中国食品药品检定研究院,硅胶g薄层板(gf254硅胶板),高效硅胶g薄层板、硅胶h薄层板购于德国默克公司,所用试剂均为分析纯。
69.1、灯心草的薄层鉴定
70.1.1、取实施例1制得的样品2.5g,研细,加甲醇20ml,超声处理20min,滤过,滤液蒸干,残渣加水20ml溶解,过固相萃取小柱,所述固相萃取小柱的规格为c18,250mg/6ml,弃去流出液,加甲醇1ml洗脱定容,作为供试品溶液;
71.1.2、取灯心草药材1g,加甲醇100ml,加热回流1小时,放冷,滤过,滤液蒸干,残渣用乙醚2ml洗涤,弃去乙醚液,加甲醇1ml使溶解,作为对照药材溶液;
72.1.3、吸取上述供试品溶液4~8μl、对照药材溶液2μl,分别点于同一硅胶g薄层板(gf254硅胶板)上,加入展开剂,所述展开剂由环己烷和乙酸乙酯按体积比(9:7)组成,预饱和后展开,取出,晾干,置紫外光灯下检视,供试品色谱中在254nm波长下可检视出与对照药材相应斑点。
73.试验结果如图1所示。
74.2、黄连的薄层鉴定
75.参考《中国药典》2020年版黄连项下,采用tlc鉴别黄连对照药材和盐酸小檗碱,其中:展开剂为:环乙烷:乙酸乙酯:异丙醇:甲醇:水:三乙胺(3:3.5:1:1.5:0.5:1)(所述展开剂各溶液混合按体积比算);薄层板为高效硅胶g薄层板,供试液点样量为1μl,对照点样量为1μl,检视条件为365nm紫外光下。
76.试验结果如图2所示。
77.3、金银花的薄层鉴定
78.参考《中国药典》2020年版金银花项下,采用tlc鉴别绿原酸,其中:展开剂为:乙酸丁酯:甲酸:水(7:2.5:2.5)(所述展开剂各溶液混合按体积比算);薄层板为硅胶h薄层板,供试液点样量为20μl,对照点样量为10μl,检视条件为365nm紫外光下。
79.试验结果如图3所示。
80.实施例3、小儿银连颗粒剂的特征图谱检测
81.2.1、色谱条件与系统适用性试验
82.以十八烷基硅烷键合硅胶为填充剂(waters acquity uplc beh c18色谱柱,50
×
2.1mm,1.7μm);以乙腈为流动相a,以含0.05%十二烷基磺酸钠的0.1%磷酸水溶液为流动相b,按表1的规定进行梯度洗脱,流速:0.4ml/min;检测波长为327nm,柱温30℃,理论板数按绿原酸峰计算应不低于3000;
83.表1特征图谱梯度洗脱表
84.时间(分钟)a(%)b(%)0~64
→
796
→
936~97
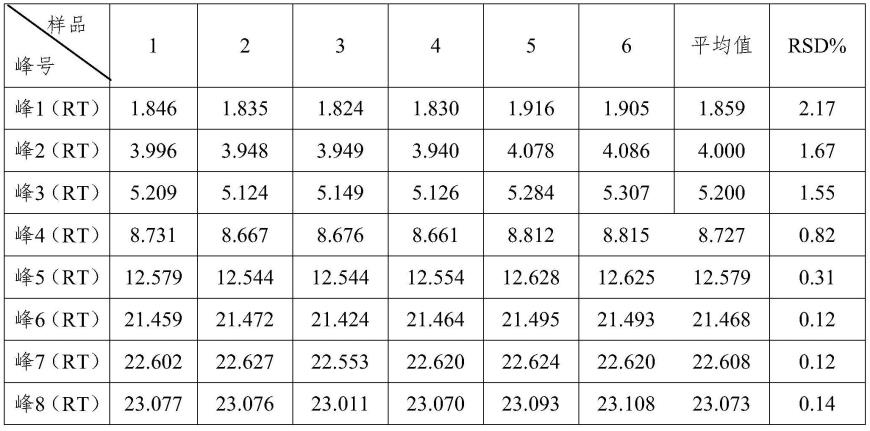
→
1093
→
909~1010
→
1690
→
8410~1616
→
2184
→
7916~1821
→
3379
→
6718~2633
→
3667
→
6426~2836
→
6064
→
40
85.2.2、对照品溶液的制备
86.取盐酸小檗碱、绿原酸对照品适量,精密称定,加甲醇制成每1ml各含40μg的溶液,得对照品溶液;
87.2.3、供试品溶液的制备
88.取实施例1制得的样品,研细,取0.5g,精密称定,置具塞锥形瓶中,精密加入50%(v/v)甲醇50ml,称定重量,超声处理30min,所述超声处理的条件为:功率250w,频率40khz,取出,放冷,再称定重量,用50%(v/v)甲醇补足减失的重量,摇匀,滤过,得供试品溶液;
89.2.4、测定法
90.分别精密吸取对照品溶液与供试品溶液各1~2μl,注入超高效液相色谱仪,测定,记录色谱图,即得。
91.试验结果如图4所示,其中:327nm检测波长时,供试品特征图谱中应呈现8个特征峰,以绿原酸为参照峰(s)峰,计算各特征峰与s的相对保留时间,其相对保留时间应在规定值的
±
7%之内。规定值:0.47(峰1)、1.00(峰2s)、1.30(峰3)、2.18(峰4)、3.14(峰5)、5.31(峰6)、5.57(峰7)、5.70(峰8),1为新绿原酸,2为绿原酸(s),3为隐绿原酸,4为阿魏酸,5为4,5-二-o-咖啡酰奎宁酸,6为黄连碱,7为巴马汀,8为盐酸小檗碱。
92.实施例4、小儿银连颗粒剂的特征图谱检测方法精密度、稳定性和重复性检测
93.2.1、色谱条件与系统适用性试验
94.以十八烷基硅烷键合硅胶为填充剂(waters acquity uplc beh c18色谱柱,50
×
2.1mm,1.7μm);以乙腈为流动相a,以含0.05%十二烷基磺酸钠的0.1%磷酸水溶液为流动相b,按表2的规定进行梯度洗脱,流速:0.4ml/min;检测波长为327nm,理论板数按绿原酸峰计算应不低于3000;
95.表2特征图谱梯度洗脱表
96.时间(分钟)a(%)b(%)0~64
→
796
→
936~97
→
1093
→
90
9~1010
→
1690
→
8410~1616
→
2184
→
7916~1821
→
3379
→
6718~2633
→
3667
→
6426~2836
→
6064
→
40
97.2.2、对照品溶液的制备
98.取盐酸小檗碱、绿原酸、阿魏酸、新绿原酸、隐绿原酸、4,5-二-o-咖啡酰奎宁酸、盐酸黄连碱、盐酸巴马汀对照品适量,精密称定,加甲醇制成每1ml各含40μg的溶液,得对照品溶液;
99.2.3、供试品溶液的制备
100.取实施例1制得的样品,研细,取0.5g,精密称定,置具塞锥形瓶中,精密加入50%(v/v)甲醇50ml,称定重量,超声处理30min,所述超声处理的条件为:功率250w,频率40khz,取出,放冷,再称定重量,用50%(v/v)甲醇补足减失的重量,摇匀,滤过,得供试品溶液;
101.2.4、测定法
102.分别精密吸取对照品溶液与供试品溶液各1~2μl,注入超高效液相色谱仪,测定,记录色谱图,即得。
103.(1)精密度试验
104.取实施例1制得的小儿银连颗粒(20201226-1),按“供试品溶液的制备”制备供试品,连续测定6次,进行精密度试验,结果见表3。
105.表3精密度试验中8个特征峰保留时间数据
[0106][0107]
由表3可知,本发明提供的小儿银连颗粒剂的特征图谱检测方法具有较高的精密度。
[0108]
(2)稳定性试验
[0109]
取实施例1制得的小儿银连颗粒(20201226-1),按“供试品溶液的制备”制备供试品溶液,精密吸取同一供试品溶液1μl,分别于制备后0,2,4,6,8,10,12h进样,依法测定,结果见下表4。
[0110]
表4稳定性试验中8个特征峰保留时间数据
[0111][0112]
由表4可知,本发明提供的小儿银连颗粒剂的特征图谱检测方法的供试品溶液在12小时内基本稳定。
[0113]
(3)重复性试验
[0114]
取实施例1制得的小儿银连颗粒(20201226-1),按“供试品溶液的制备”制备6份供试品溶液,在所确定的uplc条件下,进行测定,结果见表5。
[0115]
表5重复性试验中8个特征峰保留时间数据
[0116][0117]
由表5可知,本发明提供的小儿银连颗粒剂的特征图谱检测方法的相对标准偏差<3%,具有较好的重现性。
[0118]
(4)不同批次样品测定
[0119]
参考实施例1的制备方法提供的3批样品,按照“供试品溶液的制备”进行样品处理后,进行uplc测定,327nm检测波长下,样品图谱的相应位置应有8个特征峰,已绿原酸为参照峰(s),计算各特征峰与s峰的相对保留时间,其相对保留时间应在规定值的
±
7%之内。规定值:0.47(峰1)、1.00(峰2s)、1.30(峰3)、2.18(峰4)、3.14(峰5)、5.31(峰6)、5.57(峰7)、5.70(峰8),结果见表6。
[0120]
表6三批样品中各峰的保留时间值分析结果
[0121] 批号峰1峰2峰3峰4峰5峰6s峰7峰8120201226-10.471.001.302.183.135.355.655.76220201226-20.471.001.302.183.145.365.665.77320201226-30.461.001.302.193.165.395.695.80
[0122]
实施例5、小儿银连颗粒剂的小檗碱和绿原酸的含量测定
[0123]
3.1、色谱条件与系统适用性试验
[0124]
以十八烷基硅烷键合硅胶为填充剂(waters acquity uplc beh c18色谱柱,50
×
2.1mm,1.7μm);以乙腈为流动相a,以含0.05%十二烷基磺酸钠的0.1%磷酸水溶液为流动相b,按表7的规定进行梯度洗脱,流速:0.4ml/min;检测波长为327nm,柱温30℃,理论板数按绿原酸峰计算应不低于3000;
[0125]
表7特征图谱梯度洗脱表
[0126]
时间(分钟)a(%)b(%)0~64
→
796
→
936~97
→
1093
→
909~1010
→
1690
→
8410~1616
→
2184
→
7916~1821
→
3379
→
6718~2633
→
3667
→
6426~2836
→
6064
→
40
[0127]
3.2、对照品溶液的制备
[0128]
取盐酸小檗碱、绿原酸对照品适量,精密称定,加甲醇制成每1ml各含40μg的溶液,得对照品溶液;
[0129]
3.3、供试品溶液的制备
[0130]
取实施例1制得的样品,研细,取0.3g,精密称定,置具塞锥形瓶中,精密加入50%(v/v)甲醇50ml,称定重量,超声处理30min,所述超声处理的条件为:功率250w,频率40khz,取出,放冷,再称定重量,用50%(v/v)甲醇补足减失的重量,摇匀,滤过,得供试品溶液;
[0131]
3.4、测定法
[0132]
分别精密吸取对照品溶液与供试品溶液各1~2μl,注入液相色谱仪,测定,即得。
[0133]
(1)线性关系的考察
[0134]
a)绿原酸
[0135]
精密称取绿原酸对照品12.49mg,至25ml容量瓶中,50%(v/v)甲醇溶解并定容,分别精密吸取不同体积的对照品溶液,加50%(v/v)甲醇配成浓度为0.001、0.0250、0.0500、0.0999、0.2498、0.4996mg/ml对照品系列溶液,精密吸取1μl,注入液相色谱仪,测定,结果见表8。以对照品浓度(x)为横坐标,峰面积平均积分值(y)为纵坐标进行线性回归,得回归方程y=7.06
×
106x-3.04
×
104,绿原酸的线性范围在0.001~0.4996mg/ml(r2=0.9999)。
[0136]
表8绿原酸的线性关系考察
[0137][0138]
b)小檗碱
[0139]
精密称取盐酸小檗碱对照品12.60mg,至25ml容量瓶中,50%(v/v)甲醇溶解并定容,分别精密吸取不同体积的对照品溶液,加50%(v/v)甲醇配成浓度为0.0005、0.0126、0.0252、0.0504、0.1008、0.1512、0.2016、0.252、0.504mg/ml对照品系列溶液,精密吸取1μl,注入液相色谱仪,测定,结果见表9。以对照品浓度(x)为横坐标,峰面积平均积分值(y)为纵坐标进行线性回归,得回归方程y=6.40
×
106x+4.93
×
102,盐酸小檗碱的线性范围在0.0005~0.504mg/ml(r2=0.9999)。
[0140]
表9盐酸小檗碱的线性关系考察
[0141][0142]
(2)精密度试验
[0143]
精密量取线性关系考察实验样品,绿原酸对照品溶液(0.4996mg/ml)1.5ml,小檗碱对照品溶液(0.5040mg/ml)1.5ml,混合,50%(v/v)甲醇定容于5ml量瓶中,摇匀滤过即得,对照品溶液连续进样6次,每次1μl,记录二者的峰面积并计算rsd值,结果见表10。
[0144]
表10精密度试验结果
[0145]
测定次数绿原酸峰面积小檗碱峰面积11002466908928
2102762293868631017107925664410149429241005101375892333061015882925691峰面积均值1015296.17924399.83rsd/%0.791.02
[0146]
由表10可知,本发明提供的小儿银连颗粒剂的小檗碱和绿原酸的含量测定方法具有较高的精密度。
[0147]
(3)稳定性试验
[0148]
取实施例1制得的小儿银连颗粒(20201226-1)样品0.3g,按照“供试品溶液的制备”方法制成供试品溶液,分别于制备后0,1,2,4,6,8,10,12h进样1μl,记录小檗碱及绿原酸色谱峰的峰面积并计算rsd值。结果显示供试品溶液中的二者在12h内稳定,见表11和表12。
[0149]
表11绿原酸稳定性考察结果
[0150][0151]
表12小檗碱稳定性考察结果
[0152][0153]
(4)重复性试验
[0154]
取实施例1制得的小儿银连颗粒(20201226-1)样品0.3g,按照“供试品溶液的制备”方法制成供试品溶液,共6份,测定小檗碱和绿原酸的峰面积,计算含量及rsd值。结果见
表13,表明该方法重复性良好。
[0155]
表13重复性试验结果
[0156][0157][0158]
(5)加样回收率试验
[0159]
采用加样回收法,取实施例1制得的小儿银连颗粒(20201226-1)样品0.15g,平行6份,置具塞锥形瓶中,按100%的比例加入盐酸小檗碱和绿原酸对照品溶液,按“供试品溶液的制备”方法,测定盐酸小檗碱和绿原酸的峰面积,计算二者的含量,并按下式计算回收率。回收率=(加入对照品后含量-样品含量)/加入对照品量
×
100%
[0160]
结果显示绿原酸的平均加样回收率为100.00%,盐酸小檗碱的平均加样回收率98.03%,均符合要求,见表14和表15。
[0161]
表14绿原酸加样回收率
[0162][0163]
表15盐酸小檗碱加样回收率
[0164][0165]
(6)专属性试验
[0166]
参考实施例1处方中的药味比例,自配不含金银花的群药及不含黄连的群药,参考实施例1的制备方法制成金银花阴性对照样品和黄连阴性对照样品,按按“供试品溶液的制备”方法制成金银花阴性供试品溶液和黄连阴性供试品溶液,结果显示绿原酸和小檗碱对应的色谱峰位置无干扰。
[0167]
试验结果如图5和图6所示,说明本发明提高的小儿银连颗粒剂的小檗碱和绿原酸的含量测定具有较高的专属性。
[0168]
实施例6、小儿银连颗粒剂的绿原酸的含量测定
[0169]
3.1、色谱条件与系统适用性试验
[0170]
以十八烷基硅烷键合硅胶为填充剂(waters acquity uplc beh c18色谱柱,50
×
2.1mm,1.7μm);以乙腈为流动相a,以含0.05%十二烷基磺酸钠的0.1%磷酸水溶液为流动相b,按表16的规定进行梯度洗脱,流速:0.4ml/min;检测波长为327nm,柱温30℃,理论板数按绿原酸峰计算应不低于3000;
[0171]
表16特征图谱梯度洗脱表
[0172]
时间(分钟)a(%)b(%)0~64
→
796
→
936~97
→
1093
→
909~1010
→
1690
→
8410~1616
→
2184
→
7916~1821
→
3379
→
6718~2633
→
3667
→
6426~2836
→
6064
→
40
[0173]
3.2、对照品溶液的制备
[0174]
取绿原酸对照品适量,精密称定,加甲醇制成每1ml各含40μg的溶液,得对照品溶液;
[0175]
3.3、供试品溶液的制备
[0176]
取实施例1制得的样品,研细,取0.3g样品和稳定剂0.3mg,所述稳定剂保护剂由精氨酸、抗坏血酸钠和脂肪酸甲酯聚氧乙烯醚按1:1:2组成,精密称定,置具塞锥形瓶中,精密加入50%(v/v)甲醇50ml,称定重量,超声处理30min,所述超声处理的条件为:功率250w,频
率40khz,取出,放冷,再称定重量,用50%(v/v)甲醇补足减失的重量,摇匀,滤过,得供试品溶液;
[0177]
3.4、测定法
[0178]
分别精密吸取对照品溶液与供试品溶液各1~2μl,注入液相色谱仪,测定,即得。
[0179]
(1)稳定性试验
[0180]
取实施例1制得的小儿银连颗粒(20201226-1)样品0.3g,按照“供试品溶液的制备”方法制成供试品溶液,分别于制备后0,1,2,4,6,8,10,12h进样1μl,记录绿原酸色谱峰的峰面积并计算rsd值。结果见表17。
[0181]
表17绿原酸稳定性考察结果
[0182][0183]
(2)重复性试验
[0184]
取实施例1制得的小儿银连颗粒(20201226-1),按照“供试品溶液的制备”方法制成供试品溶液,共6份,测定绿原酸的峰面积,计算含量及rsd值。结果见表18。
[0185]
表18重复性试验结果
[0186][0187]
由表17和表18可知,添加了本发明提供的保护剂可以有效的提高小儿银连颗粒剂的绿原酸的含量测定方法的稳定性和重复性,进一步提高小儿银连颗粒剂的绿原酸的含量测定方法的准确性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1