当归四逆汤中六种成分在血浆中的浓度的检测方法与流程
1.本发明属于药物分析技术领域,特别是涉及一种当归四逆汤中六种成分在血浆中的浓度的检测方法。
背景技术:
2.当归四逆汤出自东汉张仲景的《伤寒论》,收录于《古代经典名方目录(第一批)》。全方由当归、桂枝、白芍、细辛、木通、甘草和大枣7味药组成,具有温经散寒、养血通脉等功效,是治疗血虚寒厥证的代表方剂,可用于治疗雷诺病、痛经、肩周炎、糖尿病伴随周围神经病变等疾病,具有很好的开发价值。
3.目前,现有文献报道,主要是应用高效液相色谱法测定当归四逆汤制剂部分有效成分的含量,然而,目前无法找到一种合适的液相色谱条件,可以将当归四逆汤入血后的6种成分都进行分离、检测,由于检测下限不够,只能检测当归四逆汤制剂中的几种成分的含量,不能对整个方剂在体内的入血成分进行检测,无法实现对当归四逆汤整个方剂在体内药代动力学的全面研究。
4.此外,当归四逆汤中的细辛属有毒药材,所含马兜铃酸成分具有肝、肾毒性,随着国家对经典名方的愈加重视,尤其是含有毒性药材的方剂,对其在体内入血情况的研究十分重要,既能给药效物质基础研究提供依据,又可以监测毒性成分,给经典名方的安全用药提供体内数据支持,同时检测毒性成分入血后的情况对指导当归四逆汤制剂的临床应用也是有意义的。
技术实现要素:
5.基于此,本发明的目的之一是提供一种当归四逆汤中六种成分在血浆中的浓度的检测方法,该检测方法操作简单、分析快速、特异性高、分离度高。
6.实现上述发明目的的具体技术方案包括如下:
7.一种当归四逆汤中六种成分在血浆中的浓度的检测方法,包括以下步骤:在血浆样品中加入内标工作溶液和甲醇,离心,取所得上清液作为待测血浆样品溶液;对待测血浆样品溶液进行液相色谱分离后,进行质谱分析;所述内标工作溶液为吲哚美辛溶液。
8.在其中一些实施例中,所述液相色谱分离中梯度洗脱所采用的流动相b为乙腈,流动相a为含有1.6mm~2.4mm乙酸铵的乙酸溶液,所述乙酸溶液的质量百分浓度为0.08%~0.12%。
9.在其中一些实施例中,所述梯度洗脱程序为:
10.0~0.3分钟,流动相b:40
±
5%
→
40
±
5%;
11.0.3~1.2分钟,流动相b:40
±
5%
→
95
±
5%;
12.1.2~2.0分钟,流动相b:95
±
5%
→
95
±
5%;
13.2.0~2.2分钟,流动相b:95
±
5%
→
40
±
5%;
14.2.2~3分钟,流动相b:40
±
5%
→
40
±
5%。
15.在其中一些实施例中,所述液相色谱的色谱条件包括:
16.色谱柱:以十八烷基硅烷键合硅胶为填充剂的色谱柱;
17.流速:0.4
±
0.05ml/min;
18.进样体积:5.00
±
0.5μl;
19.柱温:40
±
5℃;
20.自动进样器温度:4
±
4℃;
21.洗针液:50
±
5%乙腈水溶液;
22.洗冲速度:30μl/sec~40μl/sec;
23.洗针液体积:400μl~600μl。
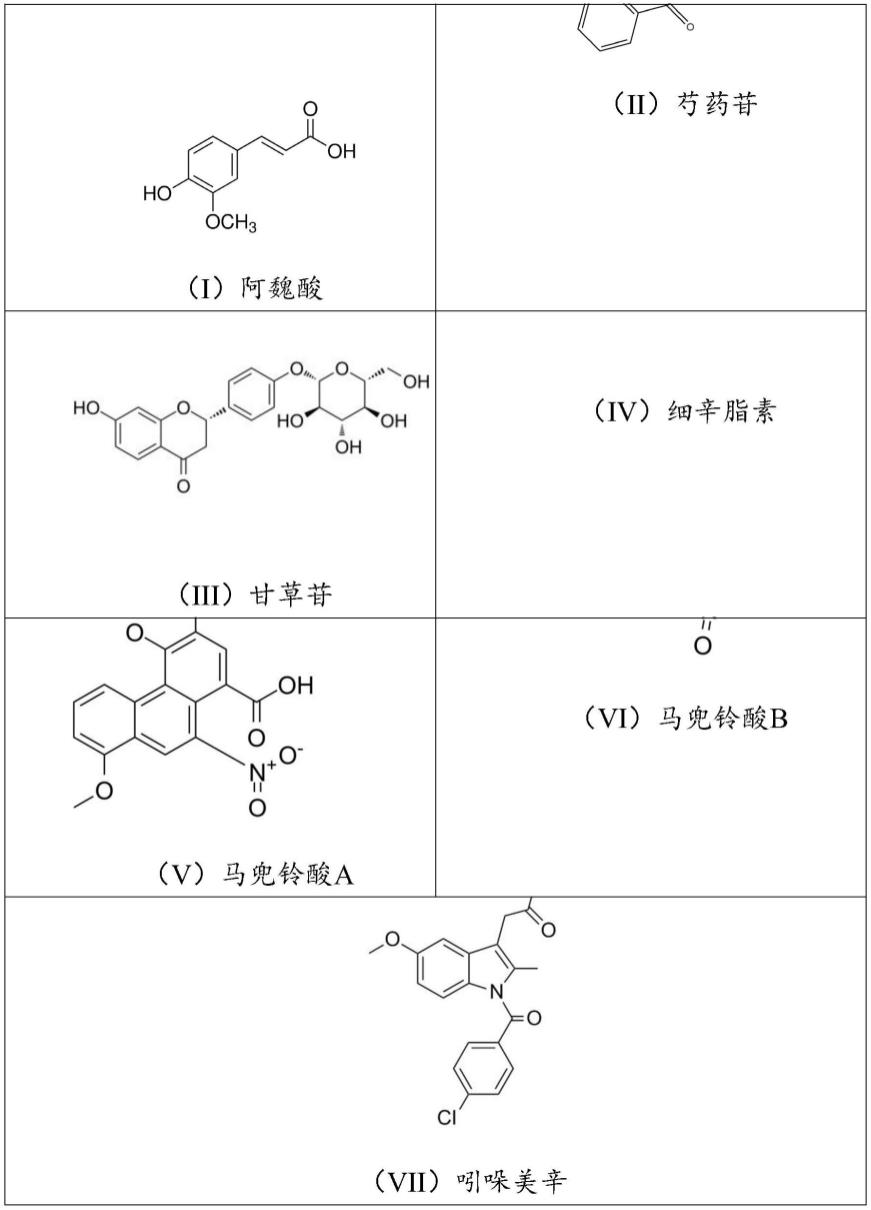
24.在其中一些实施例中,所述质谱分析的条件包括:
25.离子化模式:电喷雾离子源;正离子模式;多反应监测;
26.离子源参数:气帘气:20psi;离子源气体1:30psi;离子源气体2:30psi;
27.离子源喷雾电压:5500
±
10v;
28.离子源温度:500
±
10℃;
29.分辨率q1/q3:unit/unit;
30.碰撞气:medium;
31.暂停时间:20
±
1msec;
32.质谱采集时间为3.00
±
0.2min。
33.在其中一些实施例中,所述质谱分析中,各成分的四级杆质谱条件为:
34.阿魏酸:离子对:192.5
→
133.9;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-23.00ev;停留时间:100.0msec;
35.芍药苷:离子对:525.2
→
449.3;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-19.00ev;停留时间:100.0msec;
36.甘草苷:离子对:417.1
→
255.0;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-28.00ev;停留时间:100.0msec;
37.细辛脂素:离子对:372.2
→
233.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:12.00ev;停留时间:200.0msec;
38.马兜铃酸a:离子对:359.2
→
298.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:25.00ev;停留时间:200.0msec;
39.马兜铃酸b:离子对:329.2
→
237.8;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:25.00ev;停留时间:200.0msec;
40.吲哚美辛:离子对:358.0
→
139.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:24.00ev;停留时间:200.0msec。
41.在其中一些实施例中,采用shimadzulc 30ad液相色谱仪联合sciex qtrap5500质谱仪进行液相色谱分离及质谱分析,所采用的操作系统为analyst 1.6.3;所采用的液相色谱柱为:acquityhss t3,waters。
42.在其中一些实施例中,所述甲醇与所述血浆样品的体积比为3~6:1;和/或,所述血浆样品与内标溶液的体积比为1.5~2.5:1;和/或,所述离心温度为3℃~5℃,离心转速为2500rpm~3500rpm,离心时间为8min~12min。
43.在其中一些实施例中,所述血浆样品为人或动物的血浆样品。
44.在其中一些实施例中,还包括制备标准曲线样本和质控样本的步骤,所述制备标准曲线样本的方法为:在空白血浆中加入混合标准曲线工作溶液;所述制备质控样本的方法为:在空白血浆中加入混合质控工作溶液;所述制备混合标准曲线工作溶液和混合质控工作溶液均包括以下步骤:分别用50
±
5%甲醇水溶液梯度稀释浓度均为1.00mg/ml的阿魏酸、芍药苷、甘草苷、细辛脂素、马兜铃酸a和马兜铃酸b标准品储备溶液,即得。
45.与现有技术相比,本发明具有以下有益效果:
46.在本发明中,选择吲哚美辛作为内标物(吲哚美辛极性适中,可以兼顾六种成分的极性,能够较好地对六种化合物起到模拟分析物被基质效应影响的实际情况,更好的抵消其带来的影响)加入待测血浆样本中,再用甲醇沉淀蛋白,经离心取上清进行分析,样本预处理过程被大大简化,之后只需要极少的进样量(由于检测限低,只需要进样3μl)进行分析就可以满足定量要求;采用本发明的lc-ms/ms-mrm检测方法,可以有效检测当归四逆汤中阿魏酸、芍药苷、甘草苷、细辛脂素、马兜铃酸a、马兜铃酸b六种成分在血浆中的浓度,每个化合物的通道都只有唯一的目标峰,色谱峰形极佳,避免了其他如溶剂效应等的干扰,化合物通道相互独立,分离度高;每一个样本的进样时间为3分钟,分析快速;在空白血浆中未发现待检测的化合物峰,特异性高。因此,本发明的检测方法可实现对服用当归四逆汤后体内有效成分及毒性成分的同时监控,对推进当归四逆汤的进一步研发及安全性研究,极具现实意义。
附图说明
47.图1为阿魏酸在待测血浆样品溶液中的液质图(保留时间1.58min)。
48.图2为芍药苷在待测血浆样品溶液中的液质图(保留时间1.50min)。
49.图3为甘草苷素在待测血浆样品溶液中的液质图(保留时间1.51min)。
50.图4为细辛脂素在待测血浆样品溶液中的液质图(保留时间1.51min)。
51.图5为马兜铃酸a在待测血浆样品溶液中的液质图(保留时间1.39min)。
52.图6为马兜铃酸b在待测血浆样品溶液中的液质图(保留时间1.32min)。
53.图7为吲哚美辛在待测血浆样品溶液中的液质图(保留时间1.41min)。
54.图8为本发明试验例1中使用卡马西平作为内标溶液的检测结果。
55.图9为本发明试验例1中使用利血平作为内标溶液的检测结果。
56.图10为本发明试验例1中使用吲哚美辛作为内标溶液的检测结果。
57.图11为本发明试验例2中采用纯水作为流动相a时的检测结果。
58.图12为本发明试验例2中采用0.1%甲酸作为流动相a时的检测结果。
59.图13为本发明试验例2中采用0.1%乙酸作为流动相a时的检测结果。
60.图14为本发明试验例2中采用0.1%乙酸+2mm乙酸铵作为流动相a时的检测结果。
61.图15为本发明试验例3中采用acquitycsh c18,waters色谱柱的检测结果。
62.图16为本发明试验例3中采用acquitybeh c8,waters色谱柱的检测结果。
具体实施方式
63.为了便于理解本发明,下面将参照实施例对本发明进行更全面的描述,以下给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
64.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
65.在本发明中,提供了一种当归四逆汤中六种成分在血浆中浓度的检测方法,其中,6种成分的性质分别如下:
66.阿魏酸游离态分子式与分子量为c
10h10
o4/194.18,具体结构式如式(i)所示。
67.芍药苷游离态分子式与分子量为c
23h28o11
/480.47,具体结构式如式(ii)所示。
68.甘草苷游离态分子式与分子量为c
21h22
o9/418.39,具体结构式如式(iii)所示。
69.细辛脂素游离态分子式与分子量为c
20h18
o6/354.36,具体结构式如式(iv)所示。
70.马兜铃酸a游离态分子式与分子量为c
17h11
no7/341.28,具体结构式如式(v)所示。
71.马兜铃酸b游离态分子式与分子量为c
16
h9no6/311.25,具体结构如式(vi)所示。
72.本发明的检测方法所选用的内标为吲哚美辛(indometacin),溶于丙酮,略溶于乙醇,乙醚,氯仿和甲醇,微溶于苯,极微溶于甲苯,几乎不溶于水。游离态分子式与分子量为c
19h16
clno4/357.79,具体结构如式(vii)所示。
73.表1六种待测物和内标结构图
[0074][0075]
所述检测方法,具体包括以下步骤:
[0076]
(1)、储备溶液的制备
[0077]
取标准品阿魏酸、芍药苷、甘草苷、细辛脂素、马兜铃酸a、马兜铃酸b分别溶解于适量体积的dmso中,再加入3.5倍~4.5倍体积的甲醇,制得标准品储备溶液;将标准品储备溶液混合,得到混合标准品储备溶液;
[0078]
取吲哚美辛溶解于适量体积的dmso中,再加入3.5倍~4.5倍体积的甲醇,制得吲
哚美辛储备溶液;
[0079]
(2)、工作溶液的制备
[0080]
分别用(50
±
5)%甲醇水溶液稀释步骤(1)所述混合标准品储备溶液,制得混合标准曲线工作溶液和混合质控工作溶液;
[0081]
用(50
±
5)%甲醇水溶液稀释步骤(1)所述吲哚美辛储备溶液,制得内标工作溶液;
[0082]
在该步骤中,所述混合标准曲线工作溶液包括8个梯度,其中,阿魏酸、芍药苷、甘草苷、细辛脂素,马兜铃酸a、马兜铃酸b的浓度分别为40.0ng/ml、40.0ng/ml、20.0ng/ml、100ng/ml、100ng/ml和200ng/ml;80.0ng/ml、80.0ng/ml、40.0ng/ml、200ng/ml、200ng/ml和400ng/ml;200ng/ml、200ng/ml、100ng/ml、400ng/ml、400ng/ml和800ng/ml;400ng/ml、400ng/ml、200ng/ml、1000ng/ml、1000ng/ml和2000ng/ml;800ng/ml、800ng/ml、400ng/ml、2000ng/ml、2000ng/ml和4000ng/ml;2000ng/ml、2000ng/ml、1000ng/ml、4000ng/ml、4000ng/ml和8000ng/ml;8000ng/ml、8000ng/ml、4000ng/ml、16000ng/ml、16000ng/ml和32000ng/ml。
[0083]
在该步骤中,所述混合质控工作溶液包括定量下限质控工作溶液、低浓度质控工作溶液、中浓度质控工作溶液、高浓度质控工作溶液;其中,所述定量下限质控工作溶液中细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为40.0ng/ml、40.0ng/ml、20.0ng/ml、100ng/ml、100ng/ml和200ng/ml;所述低浓度质控工作溶液中细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为100ng/ml、100ng/ml、50.0ng/ml、250ng/ml、250ng/ml和500ng/ml;所述中浓度质控工作溶液中细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为1000ng/ml、1000ng/ml、500ng/ml、1500ng/ml、1500ng/ml和3000ng/ml;所述高浓度质控工作溶液中细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为7200ng/ml、7200ng/ml、3600ng/ml、15000ng/ml、15000ng/ml和30000ng/ml。
[0084]
(3)、待测血浆样品溶液的制备
[0085]
取血浆样品或匀浆液,加入内标工作溶液和甲醇,离心,取所得上清液作为待测血浆样品溶液;
[0086]
在该步骤中,所述甲醇与所述血浆样品或所述匀浆液的体积比为3~6:1;在样品预处理过程中,沉淀剂的量远大于血浆量,且进样量十分低,完全可以忽略溶剂效应的影响,使得分离效果极佳。所述离心温度为3℃~5℃;所述离心转速为2500rpm~3500rpm;所述离心时间为8min~12min。所述血浆为人或动物的血浆。
[0087]
在其中一个实施例中,所述待测血浆样品溶液的制备方法为:精密吸取血浆样品50μl,依次加入内标(吲哚美辛)25μl(50ng/ml)、甲醇300μl,涡旋10min,充分混匀,4℃下3000rpm离心10min,取上清液100μl,再加入100μl 0.1%乙酸+2mmol/l乙酸铵溶液,充分混匀后进样分析。
[0088]
(4)、将步骤(3)所得待测血浆样品溶液进行液相色谱分离后,再进行质谱分析。
[0089]
在本发明中,所采用的液相色谱质谱联用仪的型号为shimadzu lc 30ad液相色谱仪联合sciex qtrap 5500质谱仪,所采用的操作系统为analyst 1.6.3;本发明使用的sciex qtrap5500型质谱仪,其灵敏度较以前的仪器有了数量级的提升。
[0090]
所述液相色谱分离所采用的洗脱方式为:以(0.1
±
0.02)%乙酸+(2
±
0.4)mm乙酸铵溶液为流动相a,以乙腈为流动相b,进行梯度洗脱;所述梯度洗脱的过程为:
[0091]
0~0.3分钟,流动相b:40
±
5%
→
40
±
5%;
[0092]
0.3~1.2分钟,流动相b:40
±
5%
→
95
±
5%;
[0093]
1.2~2.0分钟,流动相b:95
±
5%
→
95
±
5%;
[0094]
2.0~2.2分钟,流动相b:95
±
5%
→
40
±
5%;
[0095]
2.2~3分钟,流动相b:40
±
5%
→
40
±
5%。
[0096]
优选地,所述梯度洗脱的过程为:
[0097]
0~0.3分钟,流动相b:40%
→
40%;
[0098]
0.3~1.2分钟,流动相b:40%
→
95%;
[0099]
1.2~2.0分钟,流动相b:95%
→
95%;
[0100]
2.0~2.2分钟,流动相b:95%
→
40%;
[0101]
2.2~3分钟,流动相b:40%
→
40%。
[0102]
在本发明中,所述液相色谱的色谱条件包括:
[0103]
色谱柱:以十八烷基硅烷键合硅胶为填充剂的色谱柱;具体为acquityhss t3,waters,其规格为1.8μm,2.1x50mm;
[0104]
流速:0.4
±
0.05ml/min;
[0105]
进样体积:5.00
±
0.5μl;
[0106]
柱温:40
±
5℃;
[0107]
自动进样器温度:4
±
4℃;
[0108]
洗针液:(50
±
5)%乙腈;
[0109]
洗冲速度:30~40μl/sec;
[0110]
洗针液体积:400~600μl。
[0111]
在本发明中,所述质谱分析的条件包括:
[0112]
离子化模式:电喷雾离子源;正离子模式和负离子模式;多反应监测;
[0113]
离子源参数:气帘气:20psi;离子源气体1:30psi;离子源气体2:30psi;离子源温度:550
±
10℃;分辨率q1/q3:unit/unit;碰撞气:medium;暂停时间:20
±
1msec;质谱采集时间为3.00
±
0.2min;正离子模式的离子源喷雾电压:5500
±
10v;负离子模式的离子源喷雾电压:-4500
±
10v;
[0114]
所述质谱分析中,各成分的四级杆质谱条件为:
[0115]
阿魏酸:离子对:192.5
→
133.9;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-23.00ev;停留时间:100.0msec;
[0116]
芍药苷:离子对:525.2
→
449.3;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-19.00ev;停留时间:100.0msec;
[0117]
甘草苷:离子对:417.1
→
255.0;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-28.00ev;停留时间:100.0msec;
[0118]
细辛脂素:离子对:372.2
→
233.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:12.00ev;停留时间:200.0msec;
[0119]
马兜铃酸a:离子对:359.2
→
298.0;去簇电压:60.0v;入口电压:10.00v;出口电
压:25.00v;碰撞能量:25.00ev;停留时间:200.0msec;
[0120]
马兜铃酸b:离子对:329.2
→
237.8;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:25.00ev;停留时间:200.0msec;
[0121]
吲哚美辛:离子对:358.0
→
139.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:24.00ev;停留时间:200.0msec。
[0122]
在以下实施例中所使用的试剂均来源于市售。
[0123]
以下结合附图和具体实施例对本发明作进一步详细的说明。
[0124]
实施例1一种当归四逆汤中六种成分在血浆中浓度的检测方法
[0125]
1、溶液的配制
[0126]
(1)、储备溶液的制备
[0127]
称取适量阿魏酸、芍药苷、甘草苷、细辛脂素,马兜铃酸a、马兜铃酸b的标准品,分别溶解于适量dmso中,再加入4倍体积的甲醇,制得浓度分别为1.00mg/ml的标准品储备溶液。
[0128]
取适量的吲哚美辛标准品,溶解于dmso中,再加入4倍体积的甲醇,制得浓度为1.00mg/ml的吲哚美辛储备溶液。
[0129]
(2)、标准曲线工作溶液的制备
[0130]
用50%甲醇水溶液将细辛脂素储备液、阿魏酸储备液、芍药苷储备液、甘草苷储备液、马兜铃酸a储备液和马兜铃酸b储备液进行稀释,得到:
[0131]
①
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为40.0ng/ml、40.0ng/ml、20.0ng/ml、100ng/ml、100ng/ml和200ng/ml的混合标准曲线工作溶液;
[0132]
②
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为80.0ng/ml、80.0ng/ml、40.0ng/ml、200ng/ml、200ng/ml和400ng/ml的混合标准曲线工作溶液;
[0133]
③
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为200ng/ml、200ng/ml、100ng/ml、400ng/ml、400ng/ml和800ng/ml的混合标准曲线样本工作溶液;
[0134]
④
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为400ng/ml、400ng/ml、200ng/ml、1000ng/ml、1000ng/ml和2000ng/ml的混合标准曲线工作溶液;
[0135]
⑤
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为800ng/ml、800ng/ml、400ng/ml、2000ng/ml、2000ng/ml和4000ng/ml的混合标准曲线工作溶液;
[0136]
⑥
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为2000ng/ml、2000ng/ml、1000ng/ml、4000ng/ml、4000ng/ml和8000ng/ml的混合标准曲线工作溶液;
[0137]
⑦
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为8000ng/ml、8000ng/ml、4000ng/ml、16000ng/ml、16000ng/ml和32000ng/ml的混合标准曲线工作溶液;
[0138]
⑧
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为10000ng/ml、10000ng/ml、5000ng/ml、20000ng/ml、20000ng/ml和40000ng/ml的混合标准曲线工作溶液。
[0139]
(3)、标准曲线血浆样本的制备
[0140]
向380μl的空白血浆中加入20.0μl上述混合标准曲线工作溶液,混合均匀,得到阿魏酸、芍药苷、甘草苷、细辛脂素,马兜铃酸a、马兜铃酸b的浓度分别为:
①
5.00ng/ml、
5.00ng/ml、1.00ng/ml、5.00ng/ml、5.00ng/ml和10.0ng/ml;
②
4.00ng/ml、4.00ng/ml、2.00ng/ml、10.0ng/ml、10.0ng/ml和20.0ng/ml;
③
10.0ng/ml、10.0ng/ml、5.00ng/ml、20.0ng/ml、20.0ng/ml和40.0ng/ml;
④
20.0ng/ml、20.0ng/ml、10.0ng/ml、50.0ng/ml、50.0ng/ml和100ng/ml;
⑤
40.0ng/ml、40.0ng/ml、20.0ng/ml、100ng/ml、100ng/ml和200ng/ml;
⑥
100ng/ml、100ng/ml、50.0ng/ml、200ng/ml、200ng/ml和400ng/ml;
⑦
400ng/ml、400ng/ml、200ng/ml、800ng/ml、800ng/ml和1600ng/ml;
⑧
500ng/ml、500ng/ml、250ng/ml、1000ng/ml、1000ng/ml和2000ng/ml的标准曲线血浆样本。
[0141]
(4)、质控工作溶液的制备
[0142]
用50%甲醇水溶液稀释吲哚美辛储备溶液,制得浓度为500ng/ml的内标工作溶液。
[0143]
用50%甲醇水溶液将细辛脂素储备液、阿魏酸储备液、芍药苷储备液、甘草苷储备液、马兜铃酸a储备液和马兜铃酸b储备液进行稀释,得到:
[0144]
①
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为40.0ng/ml、40.0ng/ml、20.0ng/ml、100ng/ml、100ng/ml和200ng/ml的混合质控工作溶液(定量下限质控);
[0145]
②
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为100ng/ml、100ng/ml、50.0ng/ml、250ng/ml、250ng/ml和500ng/ml的混合质控工作溶液(低浓度质控);
[0146]
③
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为1000ng/ml、1000ng/ml、500ng/ml、1500ng/ml、1500ng/ml和3000ng/ml的混合质控工作溶液(中浓度质控);
[0147]
④
细辛脂素、阿魏酸、芍药苷、甘草苷、马兜铃酸a和马兜铃酸b浓度分别为7200ng/ml、7200ng/ml、3600ng/ml、15000ng/ml、15000ng/ml和30000ng/ml混合质控样本工作溶液(高浓度质控);
[0148]
(5)、质控血浆样本的制备
[0149]
向380μl的空白血浆中加入20.0μl上述混合质控工作溶液,混合均匀。得到阿魏酸、芍药苷、甘草苷、细辛脂素,马兜铃酸a、马兜铃酸b的浓度分别为:
①
定量下限质控2.00ng/ml、2.00ng/ml、1.00ng/ml、5.00ng/ml、5.00ng/ml和10.0ng/ml;
②
低浓度质控5.00ng/ml、5.00ng/ml、2.50ng/ml、12.5ng/ml、12.5ng/ml和25.0ng/ml;
③
中浓度质控50.0ng/ml、50.0ng/ml、25.0ng/ml、75.0ng/ml、75.0ng/ml和150ng/ml;
④
高浓度质控360ng/ml、360ng/ml、180ng/ml、750ng/ml、750ng/ml和1500ng/ml的质控血浆样本。
[0150]
2、样本前处理
[0151]
大鼠口服给药当归四逆汤30分钟后,精密吸取大鼠血浆样本50μl,依次加入内标(吲哚美辛)25μl(500ng/ml)、甲醇300μl,涡旋10min,充分混匀,4℃下3000rpm离心10min,取上清液100μl,再加入100μl 0.1%乙酸+2mmol/l乙酸铵溶液,充分混匀。
[0152]
3、液相色谱-质谱检测
[0153]
对前处理后的大鼠血浆样本进行液相色谱-质谱检测。
[0154]
(1)、液相色谱条件
[0155]
仪器设备:shimadzu lc-30ad液相色谱系统。
[0156]
色谱柱:acquityhss t3(2.1
×
50mm,1.8μm),waters;柱温设置为40℃,自动进样器温度设置为4℃。
[0157]
流动相a:0.1%乙酸+2mmol/l乙酸铵溶液;流动相b:100%乙腈。
[0158]
总流速:0.40ml/min。
[0159]
流动相洗脱程序为:0~0.3分钟,流动相b:40%
→
40%;0.3~1.2分钟,流动相b:40%
→
95%;1.2~2.0分钟,流动相b:95%
→
95%;2.0~2.2分钟,流动相b:95%
→
40%;2.2~3分钟,流动相b:40%
→
40%。
[0160]
洗针液:50%乙腈。洗针模式为外部冲洗;冲洗速度:35μl/sec;冲洗端口液体:r1;冲洗液体积:500μl;冲洗模式:进针前后冲洗;冲洗浸润时间:0秒;冲洗方法:仅端口冲洗;冲洗时间:2秒;
[0161]
进样体积为5.00μl。
[0162]
六种化合物标准品和内标标准品的标准色谱图如图1~7所示。阿魏酸、芍药苷、甘草苷、细辛脂素,马兜铃酸a、马兜铃酸b、is(吲哚美辛)的保留时间分别为1.58min、1.50min、1.51min、1.51min、1.39min、1.32min、1.41min。
[0163]
(2)、质谱条件
[0164]
仪器设备:sciex qtrap5500质谱系统。
[0165]
离子化模式:电喷雾离子源(esi);正离子模式(positive)和负离子模式(negative);多反应监测(mrm)。
[0166]
离子源参数:气帘气(cur):20psi;离子源气体1(gas1):30psi;离子源气体2(gas2):30psi;离子源温度(tem):500℃;分辨率q1/q3(resolution q1/q3):unit/unit;碰撞气(cad):medium;暂停时间(mrpause):20msec;质谱采集时间为3.00min。正离子模式(positive)的离子源喷雾电压(is):5500v;负离子模式(negative)的离子源喷雾电压(is):-4500v。
[0167]
监测离子对四极杆参数:
[0168]
阿魏酸:离子对:192.5
→
133.9;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-23.00ev;停留时间:100.0msec;
[0169]
芍药苷:离子对:525.2
→
449.3;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-19.00ev;停留时间:100.0msec;
[0170]
甘草苷:离子对:417.1
→
255.0;去簇电压:-60.0v;入口电压:-10.00v;出口电压:-23.00v;碰撞能量:-28.00ev;停留时间:100.0msec;
[0171]
细辛脂素:离子对:372.2
→
233.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:12.00ev;停留时间:100.0msec;
[0172]
马兜铃酸a:离子对:359.2
→
298.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:25.00ev;停留时间:100.0msec;
[0173]
马兜铃酸b:离子对:329.2
→
237.8;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:25.00ev;停留时间:100.0msec;
[0174]
吲哚美辛:离子对:358.0
→
139.0;去簇电压:60.0v;入口电压:10.00v;出口电压:25.00v;碰撞能量:24.00ev;停留时间:100.0msec。
[0175]
4、检测结果
[0176]
大鼠口服给药当归四逆汤30分钟后,测得大鼠血浆样本中阿魏酸、芍药苷、甘草苷、细辛脂素、马兜铃酸a、马兜铃酸b的浓度如表2所示。
[0177]
表2大鼠血浆中6种成分的浓度检测结果
[0178]
待测物血浆样本(ng/ml)阿魏酸3.76芍药苷51.2甘草苷5.82细辛脂素8.2马兜铃酸a6.0马兜铃酸b0.0
[0179]
实施例2本发明的检测方法的方法学验证
[0180]
1、专属性
[0181]
采用实施例1的液相色谱-质谱检测条件,检测大鼠空白血浆,结果显示大鼠空白血浆中内源性杂质不干扰阿魏酸、芍药苷、甘草苷、细辛脂素、马兜铃酸a、马兜铃酸b的测定,大鼠空白血浆在六种成分处无色谱峰。
[0182]
2、最低定量限和线性范围
[0183]
大鼠血浆中:
[0184]
阿魏酸和芍药苷的最低定量限为2.00ng/ml,线性范围为5.00~500ng/ml;
[0185]
甘草苷的最低定量限为1.00ng/ml,线性范围为1.00~250ng/ml;
[0186]
细辛脂素和马兜铃酸a的最低定量限为5.00ng/ml,线性范围为5.00~1000ng/ml;
[0187]
马兜铃酸b的最低定量限为10.0ng/ml,线性范围为10.0~2000ng/ml;
[0188]
六种化合物的r2在0.9949~0.9989之间。
[0189]
3、准确度和精密度
[0190]
对于阿魏酸和芍药苷,配制低浓度质控5.00ng/ml、中浓度质控50.0ng/ml、高浓度质控360ng/ml的质控血浆样本;
[0191]
对于甘草苷,质控血浆样本的浓度分别为2.50ng/ml、25.ng/ml、180ng/ml;
[0192]
对于细辛脂素和马兜铃酸a,质控血浆样本的浓度分别为12.5ng/ml、75.0ng/ml、750ng/ml;
[0193]
对于马兜铃酸b,质控血浆样本的浓度分别为25.0ng/ml、150ng/ml、1500ng/ml。
[0194]
每一浓度6样本分析,连续测定3天,根据当日标准曲线计算qc(质控)样品浓度,求得每一浓度的re值及rsd值,具体如表3所示。
[0195]
表3 6种成分日内日间精密度结果
[0196][0197][0198]
表3结果表明:六种成分质控样本的日内和日间精密度都在-15.0%~15.0%之间,rsd在15.0%以内,符合中国药典-9012生物样品定量分析方法验证的要求。
[0199]
4、回收率
[0200]
低浓度、中浓度、高浓度质控样本分别配制6个样品,并同时提取18个不含待测物但含有内标的对照样本(混合基质)。对照样本提取完后,将在提取液中加入化合物保证与低中高提取样本理论浓度一致。待测物提取回收率的计算:r(%)=100%
×
c/s,c为qc样品中待测物的峰面积,s为对照样中待测物的峰面积,具体如表4所示。
[0201]
表4 6种成分回收率结果
[0202][0203][0204]
表4结果表明:六种成分质控样本的回收率在94.4~103.1之间,rsd都在15.0%以内,表明该方法稳定可靠。
[0205]
试验例1内标工作溶液对检测方法的影响
[0206]
该试验例中验证了内标工作溶液对检测方法的影响。
[0207]
使用的内标工作溶液分别为卡马西平、利血平和吲哚美辛,其他步骤均与实施例1
相同。
[0208]
结果如图8~图10所示。卡马西平(0.5min)和利血平(0.22min)的保留时间太早,与待测物的保留时间相差较大。而吲哚美辛的保留时间为1.41分钟,其余化合物保留时间在1.3min~1.6min之间,吲哚美辛与待测物保留时间接近但不重叠,说明吲哚美辛极性适中,可以兼顾六种成分的极性,能够较好地对六种化合物起到模拟分析物被基质效应影响的实际情况,更好的抵消其带来的影响,减少回收率的差异影响。
[0209]
试验例2液相色谱检测中的流动相对检测方法的影响
[0210]
该试验例中验证了液相色谱条件中的流动相a的选择对检测方法的影响。
[0211]
使用的流动相a分别为纯水、0.1%的甲酸水、0.1%的乙酸水,0.1%乙酸+2mmol/l乙酸铵溶液,其他步骤均与实施例1相同。
[0212]
结果分别如图11~14所示。
[0213]
采用纯水作为流动相a对于马兜铃酸a(图11中的左图)和马兜铃酸b(图11中的右图)来说,峰形交叉,故纯水作为流动相a不合适。
[0214]
与采用0.1%甲酸作为流动相a检测细辛脂素、马兜铃酸a和马兜铃酸b(图12中a-c)相比,采用0.1%乙酸作为流动相a,检测细辛脂素、马兜铃酸a和马兜铃酸b(图13中a-c)的峰形更好。
[0215]
使用0.1%乙酸+2mm乙酸铵作为流动相a,比单独使用0.1%乙酸相比,保留时间更接近,峰形更好,响应更高,六种成分的分离度更好(图14中a~f依次为细辛脂素、马兜铃酸a、马兜铃酸b、阿魏酸、芍药苷、甘草苷)。
[0216]
试验例3液相色谱柱的选择对检测方法的影响
[0217]
该试验例中验证了液相色谱条件中的色谱柱的选择对检测方法的影响。
[0218]
使用的色谱柱分别为acquitycsh c18、acquitybeh c18,waters和acquityhss t3(2.1
×
50mm,1.8μm),waters,其他步骤均与实施例1相同。
[0219]
结果如图15和图16所示。
[0220]
采用acquitycsh c18,waters色谱柱,对于细辛脂素、马兜铃酸a、马兜铃酸b来说(图15中a-c),保留时间较晚,不利于六种化合物的整体分离。
[0221]
采用acquitybeh c18,waters色谱柱,对于阿魏酸、芍药苷和甘草苷来说(图16中a-c),保留时间较早,不利于六种化合物的整体分离。
[0222]
结果表明,采用acquityhss t3(2.1
×
50mm,1.8μm),waters这个色谱柱,显著增强对极性分子的反相保留能力,减弱疏水性化合物保留,与100%水相兼容,且键合相抗流失能力强,适用于多组分、极性分布宽的样品分析。在本发明中对六种成分的分离效果很好。
[0223]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0224]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来
说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1