一种手性固定相法拆分安妥沙星对映体的方法与流程
1.本发明属于药物化学技术领域,具体涉及一种手性固定相法拆分安妥沙星对映体的方法。
背景技术:
2.左旋安妥沙星和右旋安妥沙星为对映异构体,其中左旋安妥沙星是药品盐酸安妥沙星的活性成分。而药品盐酸安妥沙星是我国拥有自主开发的一类新药,属于喹诺酮类药物,经结构改造后的安妥沙星抗菌作用不但优于氧氟沙星、环丙沙星,且氨基的引入能使安妥沙星有效降低光敏毒性,心脏毒性甚至低于莫西沙星,有较高的安全性。
3.药品盐酸安妥沙星中含单一左旋体,为了控制药品质量,有必要对盐酸安妥沙星片中右旋安妥沙星进行控制,但控制的前提是要将左旋安妥沙星和右旋安妥沙星拆分开。目前关于左旋安妥沙星、右旋安妥沙星测定的报道较少,如现有技术(洪建文,胡昌勤;左旋盐酸安妥沙星中对映体杂质的高效毛细管电泳法检测[j].分析测试学报,2007年第006期:888-890)中采用高效毛细管电泳法虽然实现了左旋安妥沙星、右旋安妥沙星对映体的的拆分,但是左旋安妥沙星、右旋安妥沙星的峰未达到基线分离,检测精密度(rsd大于2%)和检出限均比较低。因此,急需一种简单、快速、精度高的拆分左旋安妥沙星和右旋安妥沙星的方法。
技术实现要素:
[0004]
本发明的目的是提供一种手性固定相法拆分安妥沙星对映体的方法,该方法溶液配制简单、操作方便、拆分速度快、精度高,检出限低,还可用于测定右旋安妥沙星的含量,适用于盐酸安妥沙星片中右旋体的控制。
[0005]
发明人在研究本发明前,利用手性流动相添加剂法拆分了安妥沙星对映体,虽然该方法较现有技术有一定的优势,但是其手性流动相添加了较高浓度的手性氨基酸和硫酸铜,为满足分离效果需要用氢氧化钠调节ph至3.5,但该ph条件下流动相放置一段时间后容易产出沉淀,每次使用前需要重新过滤,同时,平衡时间较长,这些都容易对色谱柱造成损伤;此外,流动相中还包含较高比例有机相,不环保。而为了减少有机相的使用和色谱柱的损伤,同时减少配制流动相的繁琐操作,发明人通过使用手性固定相拆分方法代替手性流动相添加剂拆分方法,解决了上述问题。具体方案如下:本发明提供一种手性固定相法拆分安妥沙星对映体的方法,先配制左旋安妥沙星和右旋安妥沙星的混合对照品溶液,采用hplc法,以手性柱为色谱柱,以硫酸铜溶液-异丙醇为流动相进行分析检测;所述流动相中硫酸铜溶液和异丙醇的体积比为87-93:7-13。具体地,手性色谱柱选择飞诺美3126(d)-青霉胺手性色谱柱。
[0006]
本技术方案中通过优化hplc色谱条件,使用飞诺美3126(d)-青霉胺手性色谱柱实现了左旋安妥沙星和右旋安妥沙星的拆分。该手性色谱柱的固定相是反相填料通过疏水作用与手性选择配体d-青霉胺紧密结合而成,流动相中添加一定浓度的铜离子,样品分析时
通过铜离子分别与手性选择配体d-青霉胺和待测物形成可逆的金属络合物,当这种金属络合物是非对映金属络合物且稳定性有差异时,稳定性差的被先洗脱出来,稳定性好的络合物被后洗脱出来从而实现分离对映体的目的。
[0007]
进一步地,上述技术方案中,所述流动相中硫酸铜溶液和异丙醇的体积比为87-90:10-13;优选体积比为90:10。
[0008]
进一步地,上述技术方案中,所述硫酸铜溶液的浓度为2.8mmol/l。
[0009]
本技术方案中,使用硫酸铜溶液-异丙醇为流动相,制备简单,不需要调节流动相ph,且长期放置也不会析出沉淀,不会对色谱柱造成损伤。
[0010]
进一步地,上述技术方案中,hplc检测中,波长为301nm。
[0011]
进一步地,上述技术方案中,hplc检测中,柱温为30-40℃。
[0012]
进一步地,上述技术方案中,hplc检测中,进样量为20μl。
[0013]
进一步地,上述技术方案中,hplc检测中,流速为1.0 ml/min,所述色谱柱的规格为250
×
4.6mm。
[0014]
进一步地,上述技术方案中,所述混合对照品溶液的制备方法为:精密称取左旋安妥沙星对照品和右旋安妥沙星对照品,置量瓶中,加流动相溶解并稀释,配制成含左旋安妥沙星和右旋安妥沙星浓度各为1000μg/ml的对照品储备液,然后取适量对照品储备液配制成含左旋安妥沙星和右旋安妥沙星浓度各为300μg/ml的混合对照品溶液。
[0015]
本发明还提供一种手性固定相法拆分安妥沙星对映体的方法在盐酸安妥沙星片质量检测中的应用。
[0016]
进一步地,上述技术方案中,精密称取盐酸安妥沙星片样品适量,置量瓶中,加入流动相溶液超声至溶解后,过滤,得到供试品溶液,然后采用hplc法进行检测。
[0017]
相对于现有技术的有益效果:1.本发明通过优化hplc色谱条件,采用手性色谱实现了左旋安妥沙星、右旋安妥沙星的拆分,所采用的色谱条件分离效果好,干扰小,速度快,检测结果重复性好、检出限低。
[0018]
2.本发明的拆分方法不仅能够拆分左旋安妥沙星、右旋安妥沙星,还能对盐酸安妥沙星片中右旋体的含量进行测定,从而对盐酸安妥沙星片中右旋体进行严格控制,可以有效监控盐酸安妥沙星片的质量。
[0019]
3.本发明拆分方法前处理简便,后续也无需对流动相进行过滤,使用方便、环保,同时该方法具有良好的精密度、重复性,优于高效毛细管电泳法。
附图说明
[0020]
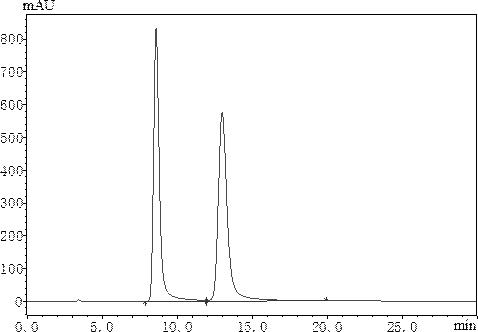
图1为本发明实施例1的色谱图,其中,左峰是右旋安妥沙星,右峰是左旋安妥沙星;图2为本发明实施例2的色谱图,其中,左峰是右旋安妥沙星,右峰是左旋安妥沙星;图3为本发明实施例3的色谱图,其中,左峰是右旋安妥沙星,右峰是左旋安妥沙星;图4为本发明实施例4的色谱图,其中,左峰是右旋安妥沙星,右峰是左旋安妥沙
星;图5为本发明实施例5的色谱图,其中,左峰是右旋安妥沙星,右峰是左旋安妥沙星;图6为本发明实施例6中供试品溶液典型色谱图,其中,左峰是右旋安妥沙星,右峰是左旋安妥沙星。
具体实施方式
[0021]
本发明的上述各项技术特征和在下文(如实施案例)中具体描述的各项技术特征之间都可以互相组合,从而构成新的或优选的技术方案,但本发明不仅仅局限于这些实施例,同样这些实施例也不以任何方式限制本发明。
[0022]
下述实施例中的实验方法,如无特别说明,均为常规方法。下述实施例涉及的制剂若无特别说明,均为普通市售品,皆可通过市场购买获得。
[0023]
下面结合图示和实施例对本发明作进一步详细描述:实验仪器和试剂:1.仪器lc-20ad xr高效液相色谱仪,带二极管阵列检测器(日本岛津公司),mettler ms205du电子天平。
[0024]
2.试剂试药右旋安妥沙星对照品(来源:中国食品药品检定研究院,批号:130466-201101,含量:90.0%),左旋安妥沙星对照品(来源:中国食品药品检定研究院,批号:130457-201101,含量:90.9%),异丙醇为色谱纯,其余试剂均为分析纯。
[0025]
盐酸安妥沙星片(生产企业:安徽环球药业股份有限公司,规格:0.1g,批号190117,平均片重0.181g,活性成分为单一安妥沙星左旋体)。
[0026]
溶液的制备:分别精密称取左旋安妥沙星对照品和右旋安妥沙星对照品27.47mg和27.58mg,置25ml量瓶中,加流动相溶解并稀释至刻度,摇匀,作为对照品储备液(分别含左旋安妥沙星和右旋安妥沙星浓度各约1000μg/ml)。
[0027]
实施例1一种手性固定相法拆分安妥沙星对映体的方法,包括以下步骤:取对照品储备液,加入流动相制备成含左旋安妥沙星和右旋安妥沙星浓度各约为300μg/ml的混合对照品溶液;按以下色谱条件进行测定:流动相:2.8mmol/l硫酸铜溶液-异丙醇(90∶10);流速:1.0ml/min;检测波长:301nm;进样量:20μl;柱温:40℃;色谱柱为飞诺美3126(d)-青霉胺手性色谱柱(250
×
4.6mm)。
[0028]
所得色谱图如图1所示,色谱参数如表1所示。
[0029]
实施例2一种手性固定相法拆分安妥沙星对映体的方法,包括以下步骤:取对照品储备液,加入流动相制备成含左旋安妥沙星和右旋安妥沙星浓度各约为300μg/ml的混合对照品溶液;按以下色谱条件进行测定:流动相:2.8mmol/l硫酸铜溶液-异丙醇(87∶13);流速:1.0ml/min;检测波长:301nm;进样量:20μl;柱温:30℃;色谱柱为飞诺美3126(d)-青霉胺手性色谱柱(250
×
4.6mm)。
[0030]
所得色谱图如图2所示,色谱参数如表1所示。
[0031]
实施例3一种手性固定相法拆分安妥沙星对映体的方法,包括以下步骤:取对照品储备液,加入流动相制备成含左旋安妥沙星和右旋安妥沙星浓度各约为300μg/ml的混合对照品溶液;按以下色谱条件进行测定:流动相:2.8mmol/l硫酸铜溶液-异丙醇(90∶10);流速:1.0ml/min;检测波长:301nm;进样量:20μl;柱温:30℃;色谱柱为飞诺美3126(d)-青霉胺手性色谱柱(250
×
4.6mm)。
[0032]
所得色谱图如图3所示,色谱参数如表1所示。
[0033]
实施例4一种手性固定相法拆分安妥沙星对映体的方法,包括以下步骤:取对照品储备液,加入流动相制备成含左旋安妥沙星和右旋安妥沙星浓度各约为300μg/ml的混合对照品溶液;按以下色谱条件进行测定:流动相:2.8mmol/l硫酸铜溶液-异丙醇(93∶7);流速:1.0ml/min;检测波长:301nm;进样量:20μl;柱温:40℃;色谱柱为飞诺美3126(d)-青霉胺手性色谱柱(250
×
4.6mm)。
[0034]
所得色谱图如图4所示,色谱参数如表1所示。
[0035]
实施例5一种手性固定相法拆分安妥沙星对映体的方法,包括以下步骤:取对照品储备液,加入流动相制备成含左旋安妥沙星和右旋安妥沙星浓度各约为
300μg/ml的混合对照品溶液;按以下色谱条件进行测定:流动相:2.8mmol/l硫酸铜溶液(含40mmol/l醋酸铵,用冰醋酸调节ph为5.0)-异丙醇(90∶10);流速:1.0ml/min;检测波长:301nm;进样量:20μl;柱温:40℃;色谱柱为飞诺美3126(d)-青霉胺手性色谱柱(250
×
4.6mm)。
[0036]
所得色谱图如图5所示,色谱参数如表1所示。
[0037]
表1 色谱参数结合图1至图5,以及表1的数据可以看出,本发明色谱条件拆分的左旋安妥沙星和右旋安妥沙星分离度好,特别是实施例1-3分析时间短,速度快,灵敏度高。
[0038]
实施例4中流动相溶液比例与实施例1不同,左旋安妥沙星和右旋安妥沙星分离度也较好,只是保留时间较最佳实施例1中增加较多,同时,右旋安妥沙星峰高下降过多,容易导致灵敏度变差。
[0039]
实施例5中在流动相中加入醋酸铵和冰醋酸,分离度和拖尾因子均较好,只是其分析时间较最佳实施例1也较长,同时由于流动相中含有较高浓度的醋酸铵和醋酸,基线噪音较大,且右旋安妥沙星峰高较实施例1中要差。
[0040]
实施例61.标准曲线的制备取左旋安妥沙星和右旋安妥沙星的对照品储备液适量,分别制备成浓度各约为1000、800、700、500、300、200、100、10、1、0.5μg/ml的混合对照品溶液,并分别按实施例1中色谱条件进行测定,并记录色谱图。以左旋安妥沙星、右旋安妥沙星的浓度(x)为横坐标,左旋安妥沙星、右旋安妥沙星的峰面积为纵坐标(y)进行线性回归。
[0041]
左旋安妥沙星浓度在约0.5~1000μg/ml之间与峰面积呈良好线性关系,回归方程为y =8.4988
×
104x
–
4.5782
×
105,r
²
= 0.9997。
[0042]
右旋安妥沙星浓度在约0.5~500μg/ml之间与峰面积呈良好线性关系,回归方程为y = 7.7719
×
104x
–
1.5223
×
105,r
²
= 0.9999。
[0043]
2.精密度试验取左旋安妥沙星、右旋安妥沙星的对照品储备液适量,制备成左旋安妥沙星、右旋安妥沙星浓度各约为300 μg/ml的混合对照品溶液,按实施例1中色谱条件进行测定并记录色谱图。混合对照品溶液连续进样6针,左旋安妥沙星、右旋安妥沙星峰面积的rsd为0.7%和0.3%,说明本方法精密度良好。
[0044]
3.定量限与检出限取左旋安妥沙星、右旋安妥沙星的对照品储备液适量,制备成左旋安妥沙星、右旋安妥沙星浓度各约为2μg/ml的混合对照品溶液,用流动相稀释成含左旋安妥沙星、右旋安妥沙星浓度各约0.5μg/ml和0.15μg/ml的溶液,分别按实施例1中色谱条件测定,记录色谱图。结果左旋安妥沙星、右旋安妥沙星峰信噪比分别满足10∶1和3∶1。本方法左旋安妥沙星、右旋安妥沙星的检出限和定量限分别为0.5μg/ml和0.15μg/ml。
[0045]
4.重复性和样品测定取盐酸安妥沙星片研细,精密称取约18mg,置10ml量瓶中,加流动相超声使溶解,过滤,作为供试品溶液,平行制备6份。精密量取上述溶液1ml,置100ml量瓶中,加流动相超声使溶解,过滤,作为对照溶液。取供试品溶液和对照溶液,分别按实施例1中色谱条件测定,并记录色谱图,其中,供试品溶液典型色谱图如图6所示。按自身对照法计算盐酸安妥沙星片中右旋安妥沙星的含量,结果为0.053%,rsd为1.0%。(其中,右旋安妥沙星含量=(供试品右旋安妥沙星峰面积/对照溶液中左旋安妥沙星峰面积)
×
100%)。
[0046]
综上所述,本发明通过优化hplc色谱条件,采用飞诺美3126(d)-青霉胺手性色谱柱实现了左旋安妥沙星、右旋安妥沙星的拆分,所采用的色谱条件分离效果好,干扰小,速度快,检测结果重复性好、精密度高、检出限低;该拆分方法还能对盐酸安妥沙星片中右旋体的含量进行测定,从而对盐酸安妥沙星片中右旋体进行严格控制,可以有效监控盐酸安妥沙星片的质量。
[0047]
最后需要强调的是,以上所述仅为本发明的优选实施例,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种变化和更改,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1