一种磷酸哌喹中三种残留溶剂含量的同时测定的方法与流程
1.本发明涉及药物检测技术领域,尤其涉及一种磷酸哌喹中三种残留溶剂含量的同时测定方法。
背景技术:
2.药品中的残留溶剂是指在原料药、辅料以及制剂生产中使用的,但在工艺生产中未能完全除去的有机挥发性化合物。在原料药合成工艺中,选择适当的溶剂可提高产量或决定药物的性质,如晶型、纯度和溶解速率。因此,溶剂在药物合成反应中是必不可少和非常关键的物质。当药品所含的残留溶剂水平高于安全值时,就会对人体或环境产生危害,因此对残留溶剂的控制已越来越受到人们的关注。
3.磷酸哌喹是治疗和控制疟疾病症的代表性药物之一,用于疟疾的治疗,也可作症状抑制性预防用,尤其是用于耐氯喹虫株所致的恶性疟的治疗与预防,亦可用于治疗矽肺;疗效确切,使用已有数十年的历史。其目前质量标准收载于《中国药典》2015年版二部,质量标准中未有残留溶剂的测定方法,使其质量控制存在缺项,无法有效的控制其质量。磷酸哌喹的合成主要是经过两次缩合反应得到1,3-双[4-(7-氯-喹啉-4-基)哌嗪-1-基]丙烷,再由1,3-双[4-(7-氯-喹啉-4-基)哌嗪-1-基]丙烷与磷酸反应制得。桂林南药股份有限公司对其的合成路线进行了改进,根据合成路线,终产品中可能残留的溶剂有甲醇、乙醇和1-溴-3-氯丙烷。
[0004]
目前对磷酸哌喹含量和有关物质的测定已有文献报道,但对磷酸哌喹原料中残留溶剂的的测定报道较少,潘勇等(潘勇,唐庆华,《液体顶空气相色谱法测定磷酸哌喹中乙醇溶剂残留》,生命科学仪器研究简报,2009年第7卷/4月刊,49-50)报道了对磷酸哌喹中乙醇溶剂残留量进行测定的方法,但该文献公开的方法仅能测定残留溶剂乙醇的含量;并未对残留溶剂1-溴-3-氯丙烷和甲醇进行含量测定。而残留溶剂1-溴-3-氯丙烷具有潜在的基因毒性杂质,需要对其残留量进行控制;甲醇对视神经影响很大,严重时可引起失明;如果不对药品中1-溴-3-氯丙烷和甲醇残留进行限度检查,将会使药品存在极大地安全隐患;且无法实现药品质量的准确控制。
技术实现要素:
[0005]
有鉴于此,本发明的目的在于提供一种磷酸哌喹中三种残留溶剂含量的同时测定方法。本发明提供的测定方法能够实现三种残留溶剂甲醇、乙醇和1-溴-3-氯丙烷的同时检测,实现了对磷酸哌喹的质量控制。
[0006]
为了实现上述发明目的,本发明提供以下技术方案:
[0007]
本发明提供了一种磷酸哌喹中三种残留溶剂含量的同时测定方法,包括以下步骤:
[0008]
将待测磷酸哌喹、乙二醇和硫酸混合,得到上机样品;
[0009]
将所述上机样品经顶空进样后,进行气相色谱检测,得到上机样品的色谱图;
[0010]
将所述上机样品的色谱图与残留溶剂的预定标准谱图进行对比,采用外标法计算得到所述待测磷酸哌喹中三种残留溶剂的含量;
[0011]
所述残留溶剂包括甲醇、乙醇和1-溴-3-氯丙烷;
[0012]
所述顶空进样的参数包括:顶空瓶的平衡温度为80~90℃,平衡时间为25~35min;
[0013]
所述气相色谱检测的参数包括:
[0014]
色谱柱为db-ffap毛细管柱、hp-ffap毛细管柱或perkinelmer elite-wax;
[0015]
色谱柱的固定液为聚乙二醇;
[0016]
分流比为5:1~20:1;
[0017]
载气为氮气;
[0018]
载气流速为5~7ml/min;
[0019]
进样量为1ml;
[0020]
升温程序为:
[0021]
在35~55℃维持10min;以50~70℃/min的速率升温至130℃,维持6min;再以50~70℃/min的速率升温至220℃,维持5min;
[0022]
进样口温度为200~240℃;
[0023]
检测器温度为200~240℃。
[0024]
优选地,所述待测磷酸哌喹、乙二醇和硫酸的用量比为0.15g:5ml:0.1ml;所述硫酸的质量浓度为95.0~98.0%。
[0025]
优选地,所述混合的方式包括超声和振摇。
[0026]
优选地,所述色谱柱的尺寸为60m
×
0.53mm
×
1.0μm、30m
×
0.53mm
×
1.0μm、30m
×
0.25mm
×
0.5μm或30m
×
0.32mm
×
0.25μm。
[0027]
优选地,所述预定残留溶剂标准谱图包括预定甲醇标准谱图、预定乙醇标准谱图和预定1-溴-3-氯丙烷标准谱图。
[0028]
本发明提供了一种磷酸哌喹中三种残留溶剂含量的同时测定方法,包括以下步骤:将待测磷酸哌喹、乙二醇和硫酸混合,得到上机样品;将所述上机样品经顶空进样后,进行气相色谱检测,得到上机样品的色谱图;将所述上机样品的色谱图与残留溶剂的预定标准谱图进行对比,采用外标法计算得到所述待测磷酸哌喹中三种残留溶剂的含量;所述残留溶剂包括甲醇、乙醇和1-溴-3-氯丙烷;所述顶空进样的参数包括:顶空瓶的平衡温度为80~90℃,平衡时间为25~35min;所述气相色谱检测的参数包括:色谱柱为db-ffap毛细管柱、hp-ffap毛细管柱或perkinelmer elite-wax;色谱柱的固定液为聚乙二醇;分流比为5:1~20:1;载气为氮气;载气流速为5~7ml/min;进样量为1ml;升温程序为:在35~55℃维持10min;以50~70℃/min的速率升温至130℃,维持6min;再以50~70℃/min的速率升温至220℃,维持5min;进样口温度为200~240℃;检测器温度为200~240℃。
[0029]
本发明的方法采用酸性(由硫酸提供)乙二醇将待测磷酸哌喹完全溶解,保证了测定结果的准确性。而且,本发明采用顶空进样法,有效地解决了直接进样法带来的样品破坏、污染衬管、进样针堵塞等问题,大大提高了工作效率。本发明提供的方法实现了磷酸哌喹中的三种残留溶剂(甲醇、乙醇和1-溴-3-氯丙烷)的同时检测,弥补了磷酸哌喹中残留溶剂检验方法的空缺,对提高磷酸哌喹的安全性有积极的作用。实施例的数据表明:本发明的
方法在考察浓度范围内的线性关系良好,回收率符合规定,灵敏度高,专属性好,结果准确,可以作为磷酸哌喹原料中残留溶剂的质量控制方法。同时,本发明的方法对1-溴-3-氯丙烷的限度为0.002%,说明本发明提供的方法灵敏度非常高。
附图说明
[0030]
图1为空白溶剂(乙二醇)的色谱图;
[0031]
图2为混合对照品溶液的色谱图;
[0032]
图3为图2的局部放大图;
[0033]
图4为实施例1中供试品溶液(样品批号为pp150102)的色谱图;
[0034]
图2~图4中,1为甲醇,2为乙醇,3为1-溴-3-氯甲烷。
具体实施方式
[0035]
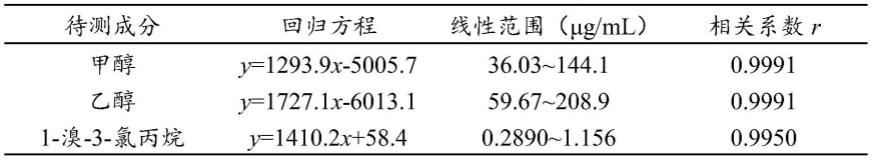
本发明提供了一种磷酸哌喹中三种残留溶剂含量的同时测定方法,包括以下步骤:
[0036]
将待测磷酸哌喹、乙二醇和硫酸混合,得到上机样品;
[0037]
将所述上机样品经顶空进样后,进行气相色谱检测,得到上机样品的色谱图;
[0038]
将所述上机样品的色谱图与残留溶剂的预定标准谱图进行对比,采用外标法计算得到所述待测磷酸哌喹中三种残留溶剂的含量;
[0039]
所述残留溶剂包括甲醇、乙醇和1-溴-3-氯丙烷;
[0040]
所述顶空进样的参数包括:顶空瓶的平衡温度为80~90℃,平衡时间为25~35min;
[0041]
所述气相色谱检测的参数包括:
[0042]
色谱柱为db-ffap毛细管柱、hp-ffap毛细管柱或perkinelmer elite-wax;
[0043]
色谱柱的固定液为聚乙二醇;
[0044]
分流比为5:1~20:1;
[0045]
载气为氮气;
[0046]
载气流速为5~7ml/min;
[0047]
进样量为1ml;
[0048]
升温程序为:
[0049]
在35~55℃维持10min;以50~70℃/min的速率升温至130℃,维持6min;再以50~70℃/min的速率升温至220℃,维持5min;
[0050]
进样口温度为200~240℃;
[0051]
检测器温度为200~240℃。
[0052]
在本发明中,如无特殊说明,本发明所用原料均优选为市售产品。
[0053]
本发明将待测磷酸哌喹、乙二醇和硫酸混合,得到上机样品。在本发明中,所述待测磷酸哌喹、乙二醇和硫酸的用量比优选为0.15g:5ml:0.1ml;所述硫酸的质量浓度优选为95.0~98.0%。在本发明中,所述混合的方式优选包括超声和振摇;本发明对所述超声和振摇的参数不做具体限定,只要能够将待测磷酸哌喹完全溶解即可。
[0054]
得到上机样品后,本发明将所述上机样品经顶空进样后,进行气相色谱检测,得到
上机样品的色谱图。
[0055]
在本发明中,所述顶空进样的参数包括:顶空瓶的平衡温度为80~90℃,优选为85℃;平衡时间为25~35min,优选为30min。
[0056]
在本发明中,所述气相色谱检测的参数包括:
[0057]
色谱柱为db-ffap毛细管柱、hp-ffap毛细管柱或perkinelmer elite-wax,优选为db-ffap毛细管柱;所述色谱柱的尺寸优选为60m
×
0.53mm
×
1.0μm、30m
×
0.53mm
×
1.0μm、30m
×
0.25mm
×
0.5μm或30m
×
0.32mm
×
0.25μm,进一步优选为60m
×
0.53mm
×
1.0μm;
[0058]
色谱柱的固定液为聚乙二醇;
[0059]
分流比为5:1~20:1,优选为10:1;
[0060]
载气为氮气;
[0061]
载气流速为5~7ml/min,优选为5ml/min;
[0062]
进样量为1ml;
[0063]
升温程序为:
[0064]
在35~55℃维持10min;以50~70℃/min的速率升温至130℃,维持6min;再以50~70℃/min的速率升温至220℃,维持5min;优选为:在45℃维持10min;以60℃/min的速率升温至130℃,维持6min;再以60℃/min的速率升温至220℃,维持5min;
[0065]
进样口温度为200~240,优选为220℃;
[0066]
检测器温度为200~240,优选为220℃。
[0067]
在本发明中,所述气相色谱检测参数能够使理论板数按甲醇峰计算不低于5000,各成分峰间的分离度均符合要求。
[0068]
得到上机样品的色谱图后,本发明将所述上机样品的色谱图与残留溶剂的预定标准谱图进行对比,采用外标法计算得到所述待测磷酸哌喹中三种残留溶剂的含量。
[0069]
在本发明中,所述残留溶剂的预定标准谱图的获取方法优选包括以下步骤:
[0070]
提供对照品溶液;
[0071]
所述对照品溶液经顶空进样后,进行气相色谱检测,得到所述残留溶剂的预定标准谱图。
[0072]
在本发明中,所述对照品溶液优选包括甲醇、乙醇和1-溴-3-氯丙烷;所述对照品溶液的溶剂优选为乙二醇;所述对照品溶液优选包括硫酸;所述硫酸的浓度和用量优选与上述技术方案一致,在此不再赘述。在本发明中,所述顶空进样和气相色谱检测的参数优选与上述技术方案一致,在此不再赘述。
[0073]
下面结合实施例对本发明提供的磷酸哌喹中三种残留溶剂含量的同时测定方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0074]
实施例1
[0075]
1溶液配制及检测
[0076]
1.1溶液配制
[0077]
(1)供试品溶液的制备:分别取三个批号的磷酸哌喹(样品1、样品2和样品3)0.15g,精密称定,置20ml顶空瓶中,精密加入乙二醇5ml、硫酸(质量浓度为95.0~98.0%)0.1ml,加盖密封,超声同时振摇使磷酸哌喹溶解,摇匀,作为供试品溶液。
[0078]
(2)对照品溶液的制备:称取甲醇0.45g、乙醇0.75g、1-溴-3-氯丙烷0.09g、精密称
定,置同一50ml量瓶中,振摇使1-溴-3-氯丙烷溶解于甲醇乙醇中,加乙二醇稀释至刻度,摇匀,作为混合对照品贮备液;精密量取混合对照品贮备液1ml,置100ml量瓶中,用乙二醇稀释至刻度,摇匀,作为混合对照品稀释液;精密量取混合对照品稀释液5ml置于20ml顶空瓶中,精密加入硫酸(质量浓度为95.0~98.0%)0.1ml,加盖密封,摇匀,作为混合对照品溶液。
[0079]
图1为空白溶剂(乙二醇)的色谱图,图2为混合对照品溶液的色谱图,图3为图2的局部放大图;图4为实施例1中供试品溶液(样品批号为pp150102)的色谱图;图2~图4中,1为甲醇,2为乙醇,3为1-溴-3-氯甲烷。从图1~4可以看出:空白溶剂对甲醇、乙醇、1-溴-3-氯甲烷的测定无干扰;各物质间的分离度均能达到基线分离。
[0080]
1.2检测参数:
[0081]
顶空进样参数:顶空瓶平衡温度为85℃,平衡时间30min;
[0082]
气相色谱检测参数:色谱柱为安捷伦db-ffap毛细管柱,60m
×
0.53mm
×
1.0μm,以聚乙二醇(peg-20m)为固定液;分流比为10:1;升温程序为:柱温在45℃维持10min,以60℃/min的速率升温至130℃,维持6min,再以60℃/min的速率升温至220℃,维持5min;进样口温度为220℃;检测器温度为220℃;载气为氮气,流速:5ml/min;进样量:1ml。
[0083]
取对照品溶液顶空进样,理论板数按甲醇峰计算应不低于5000,各成分峰间的分离度均应符合要求。
[0084]
1.3测定及计算
[0085]
取对照品溶液和供试品溶液按照检测参数部分的参数分别顶空进样和气相色谱检测,记录色谱图;按外标法以峰面积计算,甲醇、乙醇、1-溴-3-氯丙烷的残留量应不得过0.3%、0.5%、0.002%。
[0086]
2方法学验证
[0087]
2.1.线性关系考察
[0088]
配制甲醇、乙醇和1-溴-3-氯丙烷的系列浓度溶液,甲醇、乙醇和1-溴-3-氯丙烷的线性关系结果见表1。
[0089]
表1回归方程和线性关系
[0090][0091]
从表1可以看出:甲醇、乙醇和1-溴-3-氯丙烷在对应浓度范围内与峰面积呈良好的线性关系。
[0092]
2.2.精密度试验
[0093]
取对照品溶液平行测定6份,计算甲醇、乙醇、1-溴-3-氯丙烷峰面积的rsd,分别为3.60%、2.28%、1.18%(n=6),表明色谱系统精密度良好。
[0094]
2.3.重复性试验
[0095]
由于本品残留溶剂含量较低,故使用回收率项下100%回收率溶液作为重复性试验的评估,甲醇、乙醇、1-溴-3-氯甲烷的rsd分别为0.91%、0.98%、2.18%(n=6),表明重
复性良好。
[0096]
2.4检出限与定量限
[0097]
将对照品溶液逐步稀释测定,以s/n≈3确定检出限、s/n≈10确定定量限,结果甲醇、乙醇、1-溴-3-氯丙烷的检出限分别为0.088、0.13、0.068μg
·
ml-1
,定量限分别为0.31、0.39、0.23μg
·
ml-1
。
[0098]
2.5回收率的测定
[0099]
测定并计算各组分的平均回收率及rsd值,结果见表2。
[0100]
表2磷酸哌喹中残留溶剂回收率试验结果(n=9)
[0101]
[0102][0103]
从表2可以看出:本发明提供的方法回收率良好。
[0104]
2.6耐用性试验
[0105]
取对照品溶液,通过改变载气流速(6.0
±
1.0)ml
·
min-1
、进样口温度(220
±
20)
℃、分流比(5:1,20:1)、升温程序的初始温度(45
±
10)℃、升温速率(60
±
10)℃/min、检测器温度(220
±
20)℃;顶空平衡时间(30
±
5)min、加热温度(85
±
5)℃;不同色谱柱(安捷伦hp-ffap(30m
×
0.53mm
×
1.0μm)、安捷伦db-waxetr(30m
×
0.25mm
×
0.5μm)、perkinelmer elite-wax(30m
×
0.32mm
×
0.25μm)顶空进样,考察实验条件下的系统适用性。结果各实验条件下的系统适用性参数符合要求,表明该方法耐用性良好。
[0106]
2.71-溴-3-氯丙烷限度的制定
[0107]
1-溴-3-氯丙烷具有潜在的基因毒性,目前未见文献报道对其遗传毒性进行安全性风险评估,文献报道也未对其限度进行确定,本发明首次确定了其限度:根据ich m7指导原则拟定限度:磷酸哌喹按最长使用的矽肺的的防治计算,治疗量0.5g~0.75g/次,1次/周,1月量2g,半年为1疗程;间歇1月后,可进行第二疗程。也就是说7个月1个周期,1个周期服用天数为24天,那么按最保守的70年计算,总的治疗期=70
×
12/7
×
24=2880天=7.9年。因此按ich m7表2计算,1-溴-3-氯丙烷的日服剂量为0.5g,pde为10μg/天。因此,1-溴-3-氯丙烷的限度=pde/日服剂量=(10μg/天)/(0.5g/天)=20ppm,即0.002%。本发明的方法在如此低的一个限度(0.002%)可准确的测定,说明本发明的方法灵敏度非常高。
[0108]
2.8样品溶解溶剂的选择
[0109]
根据相关指导原则,测定残留溶剂样品所用溶剂应能完全溶解样品,目前已有的文献报道测定磷酸哌喹残留溶剂的方法中均未能完全溶解磷酸哌喹,本发明经大量试验,尝试使用二硫化碳、二甲苯、四氯化碳、正庚烷、甲苯、乙腈、氯仿、正己烷、二甲亚砜、n,n-二甲基甲酰胺溶解样品,结果表明:样品均不能完全溶解。
[0110]
经试验使用乙二醇与硫酸的混合物可有效地溶解样品,保证了待测磷酸哌喹中残留的有机溶剂能够完全溶解,保证了测定结果的准确性。
[0111]
2.9进样方式的选择
[0112]
目前溶剂残留测定方式主要有直接进样法和顶空进样法,目前文献报道的磷酸哌喹残留溶剂测定法一般为直接进样法,本发明使用了顶空进样法,有效地解决了直接进样法带来的样品破坏、容易污染衬管、进样针易堵等问题,大大提高了工作效率。
[0113]
3样品的测定
[0114]
取对照品溶液和供试品溶液各适量,按1.2检测参数进行测定,按照外标法计算,结果见表3。
[0115]
表3磷酸哌喹中残留溶剂测定结果(n=2)
[0116][0117]
从表3可以看出:3批磷酸哌喹样品测定结果均符合规定(甲醇限度0.3%,乙醇限度0.5%,1-溴-3-氯丙烷拟定限度0.002%)。
[0118]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人
员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1